НЕХОДЖКІНСЬКІ ЗЛОЯКІСНІ ЛІМФОМИ
Неходжкінські злоякісні лімфоми (НЗЛ) — це пухлини, що розвиваються з клітин лімфоїдної тканини і характеризуються локальним ростом. У початкових стадіях розвитку вони не супроводжуються дифузними ураженнями кісткового мозку.
НЗЛ надзвичайно гетерогенні за походженням, рівнем диференціації та функціональними ознаками клітин, з яких складається субстрат пухлини.
У дорослих у більшості випадків діагностують первинні пухлини лімфатичних вузлів різних груп. Екстранодальні осередки ураження зустрічаються рідше. У дітей нерідко спостерігаються ураження тимуса — одного з центральних органів лімфопоезу — з одночасним поширенням процесу і на регіонарні лімфатичні вузли.
Первинна локалізація процесу звичайно корелює з цитологічним варіантом пухлини та значною мірою визначає клінічні прояви захворювання і прогноз.
І серед дітей, і серед дорослих із НЗЛ переважають особи чоловічої статі. За даними зарубіжних авторів, співвідношення хлопчиків і дівчаток складає від 1,9:1 до 2,6:1. Чоловіки хворіють у 2 рази частіше, ніж жінки. Піки захворюваності спостерігаються у віці від 7 до 11 років, від 16 до ЗО років і понад 90 років. Інколи НЗЛ виявляють на 1—2-му році життя.
Етіологія. Етіологія НЗЛ дотепер остаточно не з'ясована. Підвищений ризик розвитку цих захворювань спостерігається за наявності природжених чи набутих імунодефіцитів, зокрема синдрому Віскотта—Олдріча, інфантильної (зчепленої з Х-хромосомою) агаммаглобулінемії, атаксії-телеангіек- тазії, варіабельних (некласифікованих) форм імунологічної недостатності, синдрома Чедіака—Xiraci. Певне значення може мати антигенна стимуляція при хронічних інфекційних захворюваннях, що зумовлюють дисфункцію імунорегулятор- них механізмів.
Збільшення частоти лімфом спостерігається на фоні імунодепресії після трансплантації органів, а також при таких автоімунних захворюваннях, як системний червоний вовчак, синдром Шегрена.
Описано низку лімфом, індукованих вірусами в експерименті. У хворих на Т-клітинний лейкоз був виділений вірус, що спричинює розвиток злоякісних лімфопроліферативних захворювань у людини. Є дані про зв'язок вірусу Епстейна— Барр з лімфомою Беркітта, яка зустрічається в дітей африканського континенту, де вона є ендемічною. У сироватці крові дітей з лімфомою Беркітта виявлено високий титр противірусних антитіл.
Встановлено, що вірус Епстейна—Барр має тропізм до нормальних В-лімфоцитів і може спричинювати трансформацію В-клітин in vitro.
Завдяки досягненням цитогенетики та молекулярної біології були отримані нові дані про патогенез деяких форм НЗЛ. У клітинах лімфоми Беркітта та інших лімфом, діагностованих у хворих у неендемічних районах, були виявлені набуті аномалії каріотипу. При цьому генетичний матеріал дистального сегмента довгого плеча 8-ї хромосоми трансло- кується на 14-ту хромосому, де розташовується локус легких каппа-ланцюгів, та на 22-гу хромосому, де розташовується локус лямбда-ланцюгів.
Доведено, що людський гомолог онкогена вірусу мієлоци- томатозу птахів (MC 29), який називається с-тус і локалізується на 8-й хромосомі, у клітинних лініях лімфоми Беркітта також транслокується на 14-ту хромосому. Структура генетичного матеріалу при такій транслокації сприяє рекомбінації ділянки 5'-πpoτo-myc з ділянкою 5'-гена імуногло- буліну. Однак транслокації, що пов'язані з НЗЛ, не завжди включають локус прото-тус. У клітинах деяких «не- беркіттівських» лімфом імуноглобуліновий локус 14-ї хромосоми рекомбінує з невідомими локусами 18-ї хромосоми.
Останнім часом у клітинах лінії лімфоми Беркітта з t(8; 22) була виявлена транслокація на 8-му хромосому та експресія протоонкогена c-sis. Відомо, що його експресія в нормальних диференційованих клітинах дуже обмежена. Ці дані узгоджуються з гіпотезою про багатостадійність канцерогенезу та активацію низки онкогенів, що при цьому відбувається.
Таким чином, зміни регуляції або експресії генів, що пов'язані з процесами проліферації та диференціювання, відіграють важливу роль у патогенезі лімфоїдних новоутворень.
Можливо, саме вони і блокують диференціювання клітин.Периферичні органи імунної системи зазнають впливу факторів зовнішнього середовища з перших днів після народження дитини, а можливо, і в антенатальний період, і реагують на дію численних антигенних і мітогенних факторів проліферативною реакцією.
Підвищення проліферативної активності при повторних впливах збільшує ризик виникнення генетичних порушень у певних стадіях диференціювання тієї чи іншої субпопуляції клітин.
Спектр лімфоїдних пухлин у дітей відрізняється від такого, що зустрічається в дорослих, оскільки в дітей пухлини виникають з ранніх лімфоїдних клітин-попередників, а в дорослих — зі спеціалізованих імунокомпетентних клітин в антигенза- лежній стадії диференціювання. Цим можна пояснити той факт, що в дітей надзвичайно рідко зустрічаються пухлини, що походять із клітин зародкових центрів лімфоїдних фолікулів, і ніколи не розвиваються високодиференційовані лімфоцитарні лімфоми.
Гістологічна і цитологічна класифікація. Уявлення про НЗЛ неодноразово змінювалися залежно від трактування природи клітин, що складають субстрат пухлини. Тому було запропоновано велику кількість класифікацій НЗЛ.
Найбільшого поширення набули такі класифікації:
Lukes — Collins (1976);
кільська (1974) з модифікаціями (1976, 1988, 1989); британська (1974);
класифікація, що була рекомендована ВООЗ (1966); робоча класифікація (Working Formulation, 1982).
Однак усі ці класифікації мають певні недоліки (різноманітність назв пухлин одного типу, послідовність етапів і маркерів диференціювання клітин у тимусзалежних і тимус - незалежних зонах периферичних органів лімфопоезу та ін.).
Останніми роками значно розширились уявлення про природу лімфоїдних пухлин. Методи імунофенотипування і молекулярно-генетичні методи дозволили виявити нові види НЗЛ, які раніше не входили в класифікації. Міжнародна група по вивченню лімфом розробила пропозиції щодо об'єднання раціональних аспектів європейських і американських класифікацій.
Цей документ був названий «Переглянута євро- американська класифікація лімфом» (REAL-класифікація).Відповідно до REAL-класифікації пухлини лімфоїдного походження поділяють на три головні групи: 1) В-клітинні; 2) Т-клітинні; 3) хвороба Ходжкіна.
Деякі форми НЗЛ неможливо віднести до цих груп, тому їх віднесли до некласифікованих.
В- і Т-клітинні лімфоми поділяють на дві великі групи: пухлини, що походять із клітин-попередників; пухлини, що походять із клітин, які мають «периферичний» фенотип і включають решту лімфом і лейкозів.
REAL-класифікація лімфоїдних неоплазій

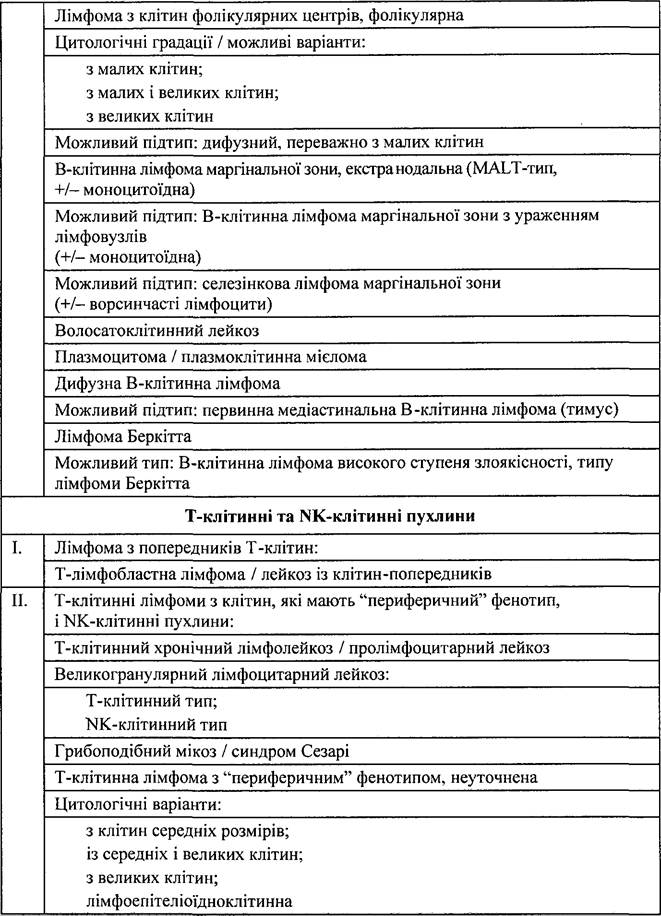

Наведена систематизація лімфом була покладена в основу класифікації ВООЗ (1995).
Класифікація пухлин з лімфоїдної тканини (ВООЗ, 1995)


Діагностика НЗЛ Грунтується на даних біопсії, гістологічних і цитологічних (у тому числі цитохімічних та імуно- фенотипових) досліджень видаленої пухлини або її частини. Обмежуватись одним діагностичним методом неприпустимо. Необхідно також провести дослідження кісткового мозку та трепанобіоптату. Для уточнення клінічної стадії захворювання, тобто поширення пухлинного процесу, проводять УЗД, комп'ютерну томографію, MPT, сцинтиграфію печінки, селезінки та кісток скелета.
Патологічні ознаки клітин. Для ідентифікації природи і ступеня диференціювання клітин використовують ци- тохімічні та імунофенотипові методи. Досліджують мазки периферичної крові і лейкоконцентратів венозної крові, відбитки видалених лімфатичних вузлів та кріостатні зрізи.
Пухлини з В-клітин
Пухлини з В-клітин-попередників.
Лімфобластний / лейкоз / лімфома з В-клітин-попередників характеризується мономорф- ним клітинним складом. У відбитках лімфатичних вузлів виявляють бластні клітини округлої або овальної форми з вузьким обідком помірно базофільної цитоплазми, яка не містить зернистості. Ядерний хроматин дрібногранулярний або має більш щільну структуру. В ядрах містяться великі ядерця. За розмірами клітини поділяють на мікро- і мезогенерації (за аналогією з L1- і Ь2-типами лімфобластів при гострому лімфобластному лейкозі — ГЛЛ).У лімфобластах В-клітинної природи пероксидаза відсутня, PAS-реакція гранулярна в частині клітин. Кислу фосфатазу виявляють у невеликому відсотку клітин, кисла неспецифічна естераза відсутня.
Імунофенотипові ознаки В-лімфобластів. В-клітинні лімфо- ми в дітей, за даними різних авторів, зустрічаються в 30—40 % випадків. У дорослих В-фенотипова природа лімфобластних НЗЛ підтверджена у більшості випадків.
Клітини постійно експресують Іа-подібний антиген, а також пан-В-лімфоцитарні антигени TdT+, HLA-DR+, CD19+, CD20+, CD22+, sig—. На більшості клітин виявляють антигени CD79a і CDlO, рецептор CD3d, а також антигени CD24 і CD34. CD34 вважають антигеном стовбурових клітин. Інколи спостерігається експресія антигенів CD13 і (або) CD33.
Пухлини з периферичних В-клітин. Лімфома з малих лімфо-
53і цитів. Субстратом пухлини є клітини, подібні до малих лімфоцитів при ХЛЛ. Крім того, у відбитках лімфатичних вузлів виявляють багато лімфобластів і пролімфоцитів. У деяких клітинах спостерігаються ознаки плазматизації. Пухлинні клітини при НЗЛ із малих лімфоцитів походять із В-клітин. Вони експресують пан-В-клітинні антигени CD19 і CD20, Іа-подібний антиген. Характерною є наявність антигенів CDlO і CDlla, при цьому експресія антигенів CD5 і CD23 дуже низька. Дуже високою є щільність поверхневих імуноглобулінів (sig). Клітини не утворюють M-РУК.
Лімфома з клітин центрів фолікулів (фолікулярна). При фолікулярній лімфомі виявляють поліморфні клітини: дрібні з невеликими ядрами і конденсованим хроматином і більші за розмірами клітини з великою цитоплазмою.
Клітини можуть бути округлими. Нуклеоли майже не візуалізуються. Ядра клітин здебільшого мають глибокі вирізки, або фісури. Найчастіше субстрат пухлини представлений дрібними клітинами центрів фолікулів і центробластами (великими клітинами з центрів фолікулів з нерозщепленим ядром).Імунофенотипові ознаки. Клітини з центрів фолікулів характеризуються високою щільністю поверхневих імуноглобулінів, найчастіше slgM, або в поєднанні з slgD. Ці клітини експресують В-клітинні антигени CD19, CD20, CD79a і CD22, а також CDlO+/-, CDlla+/-, CD23-/+, CD5-.
Лімфошазмощтіарна лімфома. При лімфоплазмоцитарних НЗЛ субстрат пухлини представлений малими лімфоцитами, лімфоплазмоцитарними елементами і плазматичними клітинами. Невеликий відсоток клітин слабко експресують пан-В- клітинні антигени CD19, CD20, CD79a і CD22. Найхарактернішими фенотиповими маркерами цієї форми НЗЛ є антигени CDlO, CD22, CD38. Інколи спостерігається експресія антигенів CD25+/-, CDllc+/- при CD5-.
Цитогенетичні особливості В-клітинних НЗЛ із периферичних В-клітин. Цитогенетичні аномалії хромосом виявляють більш як у 50 % хворих із пухлинами низького ступеня злоякісності. За допомогою методу флуоресцентної гібридизації in situ (FISH) такі аномалії можна виявити в більшості хворих.
При НЗЛ із малих лімфоцитів з нескрученими ядрами найчастіше виявляють t (8, 14), (q24, q32), а також t(2, 8) або t (8, 22).
При НЗЛ із малих лімфоцитів мантійної зони виявляють транслокації з місцем розриву 14q32, де розміщується локус важких ланцюгів імуноглобулінів. Найчастіше зустрічаються транслокації t(ll, 14), (ql3; q32), але можуть спостерігатися й інші аномалії. При фолікулярній (нодулярній) лімфомі дуже часто (у 70—80 % випадків) виявляють транслокацію t(14, 18), (q32; q21).
Лімфома з клітин мантійної зони. Пухлина складається з малих або середніх розмірів лімфоідних клітин. Під час гістологічного дослідження ядра клітин здебільшого неправильної форми або розщеплені. Злоякісні клітини розміщуються в мантійній зоні фолікулів. Під час імунофенотипування виявляють В-клітинні антигени CD19, CD20, CD79a і CD22, а також антигени CD5, CDlO+/-, CD23—, CD43+. На поверхневих мембранах пухлинних клітин визначаються IgM та IgD.
В-клітинна лімфома маргінальної зони. Цей тип лімфоми характеризується клітинною гетерогенністю. Крім клітин маргінальної зони, що представлені малими атиповими клітинами, виявляють В-клітини з моноцитоідними рисами, малі ворсинчасті лімфоцити і плазматичні клітини, а також невелику кількість імунобластоподібних клітин.
Таблиця 34. Імуногістологічні і цитогенетичні ознаки В-клітинних НЗЛ із периферичних В-клітин
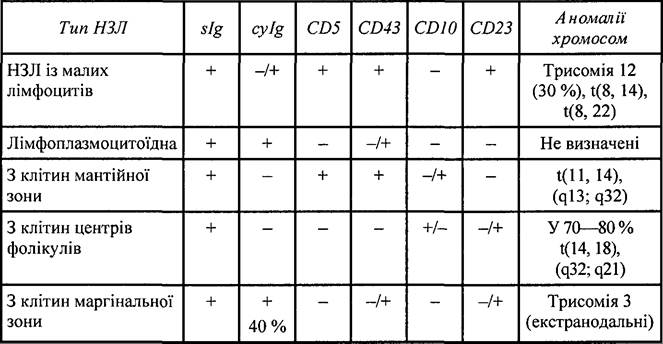
Імунофенотипові ознаки. При взаємодії з MKAT спостерігається позитивна реакція на такі антигени: CD19+, CD20+, CD22+, CD79a+, CD5-, CD23-, CD43-/+, CDll+/-, CDlO-, інколи CD25+.
Цитогенетичні ознаки. Описано трисомію 3 і транслокації t(ll, 18). Реаранжування генів bcl-2 або bcl-1 не спостерігається. Мутація відбувається в ділянці короткого плеча 9-ї хромосоми в локусі, що кодує р 16.
Дифузна В-крупноклітинна лімфома. Під час цитологічного дослідження виявляють великі базофільні клітини. Цитоплазма темно-синього кольору, широка, ці клітини нагадують бласттрансформовані різними мітогенами клітини в культурі тканини. Ядра бластів овальні або округлі, містять великі ядерця. Структура хроматину ніжнопетлиста. Від 50 до 60 % імунобластних (центробластних) лімфом виникають із В-клітин і менш як 10 % — з Т-клітин.
Під час цитохімічного дослідження імунобластів виявляють дифузну та помірну PAS-реакцію. У деяких клітинах гранули глікогену розміщуються по периферії цитоплазми.
Імунофенотип: CD19+, CD20+, CD22+, CD79a+, CD5—/+, sig+/-, cylg-/+, CD45+/—, CDll+/-, CDlO-/+.
Цитогенетичні ознаки: реаранжування гена bcl-2 унаслідок t(14, 18) у пухлинних клітинах спостерігається в ЗО % випадків. В окремих випадках виявляють перебудову с-тус. В 1/3 випадків відбуваються реаранжування та інактивація гена bcl-6.
Підтип В-великоклітинних НЗЛ — первинна лімфома середостіння (тимуса). Субстрат пухлини представлений великими клітинами, які нагадують центробласти, і великими центроцитами. Деякі клітини подібні до імунобластів і клітин Бере- зовського—Штернберга.
Імунофенотип: CD19+, CD20+, CD22+, CD79a+, cylg—, CD45+/—.
Перебіг захворювання агресивний, часто розвиваються екстранодальні рецидиви. Уражуються печінка та інші органи травлення, а також ЦНС. Пухлини тимуса частіше виникають у молодих жінок.
Лімфома Беркітта. У відбитках і пунктатах пухлин, а також в асцитичній рідині переважають бластні клітини середніх розмірів, округлої форми, з дуже базофільною вакуолізова- ною цитоплазмою. Ядра клітин округлі чи овальні. Структура хроматину груба. За FAB-класифікацією ГЛЛ, бласти відносять до L3.
Цитохімічні дослідження свідчать, що в беркіттоподібних бластах PAS-реакція невиражена, дифузна. Реакція на кислу фосфатазу також дифузна, реакція на кислу неспецифічну ес- теразу — негативна.
Під час імунологічного дослідження виявляють експресію IgM на поверхневих мембранах, виражену експресію Іа-подібного антигену та пан-В-клітинних антигенів. Пухлинні клітини реагують із MKAT FMC-7, FMC-8. Вони слабко реагують із FMC-L На відміну від справжньої лімфоми Беркітта, СЗ і Fc-рецептори, а також рецептори до вірусу En- стейна—Барр не виявляють.
Цитогенетичні ознаки. У більшості випадків спостерігається транслокація онкогена c-myc, локалізованого на 8-й хромосомі, у зону важкого ланцюга імуноглобуліну.
При беркіттоподібній НЗЛ частіше виявляють перебудову гена bcl-2.
Пухлини з Т-і NK-клітин
Пухлини з Т-клітин-попередників. Лімфобластний лейкоз / лімфома з Т-клітин-попередників. Пухлинні клітини представлені малими або великими лімфобластами з округлими або конвалютивними ядрами, «ніжним» хроматином і вузьким обідком цитоплазми. При забарвлюванні клітини не відрізняються від В-лімфобластів.
Цитохімічні ознаки. Реакція на пероксидазу негативна. PAS-реакція позитивна, гранулярна. Надійний ідентифікаційний тест — цитохімічне визначення активності кислої фосфатази в Т-лімфобластах. Продуктами реакції є гранули, які виявляють у цитоплазмі. Подібна реакція спостерігається і при визначенні активності кислої неспецифічної естерази.
Імунофенотип. У більшості випадків при Т-лімфобластних лімфомах виявляють антигени CD7+, CD3+, CD2+/—, CD5—/+, CDla+/-. На поверхневих мембранах і цитоплазмі імуноглобуліни не виявляють.
Цшпогенетичні ознаки. Спостерігається реаранжування генів IgM і TCR.
Пухлини з периферичних T- і NK-клітин. Грибоподібний мікоз (синдром Сезарі). Клітини Сезарі можуть бути великими і малими. Великі клітини за розмірами такі, як гранулоцити та моноцити. Малі клітини нагадують малі лімфоцити. Вони не містять ядерець чи нуклеол, а їх ядра мають «мозкоподібну» структуру.
Під час цитохімічного дослідження виявляють позитивну реакцію на кислу фосфатазу, що характерно для Т-клітин. PAS-реакція в клітинах Сезарі гранулярна (виявляють дрібні та середні за розмірами гранули).
Імунофенотип. Клітини Сезарі мають маркери пан- Т-клітинних антигенів: CD2, CD3, CD5. Вони рідко експресують CD7, CD4+ і CD8+, частіше — CD4+, CD8—. Дуже рідко спостерігається експресія Іа-подібного (HLA-DR) антигену та CD25-pe∏eπτopa до інтерлейкіну-2.
Периферичні (невизначені) Т-клітинні лімфоми. Субстрат пухлини представлений малими і великими Т-клітинами. Спостерігається також інфільтрація незлоякісними клітинами — еозинофілами та епітеліоїдними гістіоцитами. До периферичних Т-клітинних лімфом належить лімфоепітеліоїдна лімфома (лімфома Леннерта). Серед пухлинних клітин, різних за розмірами і формою ядер, зустрічаються клітини, що нагадують клітини Березовського—Штернберга.
Цитохімічні ознаки. Спостерігається позитивна гранулярна реакція на кислу фосфатазу, кислу неспецифічну естеразу, В-глюкуронідазу. PAS-реакція гранулярна.
Імунофенотип. Клітини експресують антигени CD3, CD2, CD5, CD7. У більшості клітин виявляють антигени CD4+, але деякі пухлинні клітини експресують антигени CD4— і CD8—.
Ангіоімунобластна Т-клітинна лімфома. Пухлинні лімфоїдні клітини представлені малими лімфоцитами, імунобластами і атиповими світлими клітинами з великою блідою цитоплазмою. Крім того, зустрічаються епітеліоїдні і плазматичні клітини, а також еозинофіли.
Гістологічна структура лімфатичного вузла змінена. Синуси розширені, реактивних фолікул із зародковими центрами немає. Пухлинні клітини інфільтрують капсулу лімфатичного вузла і проникають у жирову клітковину.
Імунофенотип. Пухлинні клітини експресують антигени Т-лімфоцитів, частіше CD4+.
Цитогенетичні ознаки. Спостерігається перебудова генів TCR (Т-клітинного рецептора). У 50 % випадків виявляють рецептори до IgM. Можлива трисомія 3 або 5.
Ангіоцентрична лімфома. Пухлина складається з малих лімфоцитів, атипових лімфоїдних клітин та імунобластів. Серед останніх виявляють плазматичні клітини, еозинофіли і гістіоцити. Пухлина інфільтрує стінки судин, що призводить до закупорки просвіту судин і розвитку осередків некрозу як у пухлинній, так і в неураженій пухлиною тканині.
Імунофенотип. Пухлинні клітини мають фенотип уніпо- тентних попередників ПКК. У цитоплазмі спостерігається експресія антигену CD3, а на поверхневих мембранах — антигенів CD2, CD7 і CD56.
Т-клітинна лімфома кишечника. Раніше це захворювання мало назву «злоякісний гістіоцитоз кишечнику». Пухлина складається з малих і середнього розміру лімфоїдних клітин, а також великих анапластичних клітин і реактивно змінених гістіоцитів.
Імунофенотип. Клітини пухлини експресують антигени CD3+, CD7+, CD8+ і CD4—. Характерним є наявність CD103 (МЬА)-антигену.
Цитогенетичні ознаки. Спостерігається клональна перебудова генів р-ланцюга TCR.
Анапластична великоклітинна лімфома. Субстрат пухлини представлений великими клітинами з поліморфними ядрами, які містять численні ядерця. Зустрічаються багатоядерні клітини, які нагадують клітини Березовського — Штернберга. Пухлинні клітини при анапластичній НЗЛ значно більші, ніж при інших великоклітинних НЗЛ. Клітини пухлини зазвичай ростуть у вигляді пластів. Пухлинний процес поширюється на синуси лімфатичних вузлів.
Цитохімічні ознаки. PAS-реакція в пухлинних клітинах слабко дифузна (з гранулами в цитоплазмі окремих клітин). Активність кислої фозфатази, кислої неспецифічної естерази також незначна.
Імунофенотип. Пухлинні клітини експресують антигени CD30+ і CD68—. Можлива експресія інших Т-клітинних антигенів: CD25+/—, CMA+/-, CD45+/—, CD3+/-, CD15+/- та ін.
Цитогенетичні ознаки. Майже в 50 % випадків виявляють перебудову генів імуноглобулінів і TCR, іноді — t(2; 5).
Клініка різних форм НЗЛ залежить від морфологічного варіанту лімфоми та імунологічних особливостей T- і В-клітин.
Першим симптомом захворювання може бути збільшення лімфатичного вузла, селезінки, яєчка чи частки щитоподібної залози. Лімфатичні вузли мають щільну консистенцію, рухливі, неболючі.
У деяких випадках першим клінічним проявом захворювання є інтоксикація. Нерідко хвороба може розвиватися під маскою автоімунної гемолітичної анемії, геморагічного васкуліту, поліартриту, екземи.
Лімфатичні вузли спочатку м'які, пізніше вони стають більш щільними та утворюють конгломерати, що досягають великих розмірів (до 16—20 см у діаметрі). Вони обростають великі судини грудної порожнини і трахеї, здавлюють їх. Пухлина порушує функцію дихання або зумовлює застій у системі верхньої порожнистої вени. Може виникати набряк верхньої половини тіла (голови, рук, тулуба). Можливий розвиток багатьох судинних колатералей. Унаслідок збільшення брижових та заочеревинних лімфатичних вузлів можуть порушуватися функції кишок і сечових органів. У разі збільшення лімфовузлів у воротах печінки та здавлювання загальної жовчної протоки розвивається жовтяниця.
У більшості випадків лейкоцитарна формула не змінена. У разі прогресування процесу розвиваються нормохромна анемія і тромбоцитопенія. Зміна показників периферичної крові може бути зумовлена застосуванням цитостатичної та променевої терапії. HIOE буває як підвищеною, так і нормальною.
Клінічні особливості окремих форм НЗЛ. Лімфобластна лімфома з В-клітин-попередників.
Серед лімфобластних лімфом пухлини В-клітинного походження складають близько 20 %. Частіше хворіють діти, ніж дорослі.
Головні осередки ураження локалізуються, як і при Т-клітинних формах, у лімфатичних вузлах, шкірі, кістках.
Пухлинний процес швидко поширюється на кістковий мозок.
Відсутність на мембранах клітин антигенів CDlO, CD34 і CD24 або наявність антигенів CD13 і CD33 вважають несприятливою прогностичною ознакою при злоякісних лімфобласт- них лімфомах.
Лімфоплазмоцитарна лімфома (імуноцитома) частіше зустрічається у хворих літнього віку. Уражуються лімфатичні вузли, селезінка, кістковий мозок. Можливий розвиток і екс- транодальних осередків ураження. Пухлинні клітини можуть з'явитися також у периферичній крові. У сироватці крові виявляють моноклоновий парапротеїн типу IgM, що може зумовити розвиток синдрому гіперв'язкості.
Для лімфоми мантійної зони характерні лімфаденопатія і спленомегалія, екстранодальні ураження, особливо органів травлення. Перебіг захворювання помірноагресивний. У разі прогресування процесу спостерігається інфільтрація кісткового мозку пухлинними клітинами.
Фолікулярна лімфома з клітин центрів фолікулів (центро- цитів) зустрічається найчастіше. Як правило, хворіють дорослі. Осередки ураження виявляють у лімфатичних вузлах, селезінці, кістковому мозку. Розвиваються екстранодальні ураження. У разі прогресування захворювання можлива його трансформація в крупноклітинну В-НЗЛ.
В-клітинна екстранодальна лімфома маргінальної зони зустрічається тільки в дорослих, частіше в жінок. В анамнезі багатьох хворих є дані про синдром Шегрена, тиреоїдит Xa- шимото. При екстранодальній злоякісній лімфомі у більшості випадків виявляють ураження залозистих епітеліальних тканин різної локалізації. Перебіг захворювань тривалий, але в ЗО % випадків відбувається генералізація процесу та трансформація в крупноклітинну лімфому.
Класична лімфома Беркітта була виявлена в дітей Східної Африки та Нової Гвінеї. Захворювання характеризується ураженням кісток, лімфатичних вузлів, нирок, яєчників, легень. Кістковий мозок уражується рідко. Беркіттоподібна НЗЛ зустрічається частіше в дітей. Захворювання супроводжується ураженням абдомінальних лімфатичних вузлів (дистальних відділів тонкої кишки). Воно часто ускладнюється асцитом. У дітей пухлина характеризується дифузним ростом і екстрано- дальною локалізацією. Спостерігається ураження яєчок, яєчників, легень, нирок, кісток нижньої щелепи. У деяких випадках першим уражується кістковий мозок. Пухлинні клітини з'являються в периферичній крові. У стадії лей- кемізацїї беркіттоподібну лімфому діагностують за ФАВ-кла- сифікацією як ГЛЛ L3.
Т-лімфобластні НЗЛ зустрічаються частіше в підлітків і молодих чоловіків. Спостерігаються збільшення лімфатичних вузлів і (або) розвиток пухлини середостіння. Часто виявляють лейкемізацію.
У хворих на грибоподібний мікоз (синдром Сезарі) на шкірі з'являються бляшки. Крім того, спостерігається ураження лімфатичних вузлів.
Периферичні невизначені Т-клітинні лімфоми складають близько 15 % від усіх НЗЛ. Хворіють переважно дорослі. Перебіг хвороби агресивний. У більшості хворих виявляють ураження лімфатичних вузлів, шкіри, печінки, селезінки та інших внутрішніх органів.
Ангіоімунобластна Т-клітинна лімфома проявляється гене- ралізованою лімфаденопатією, гарячкою, зниженням маси тіла, ураженням шкіри.
Т-клітинна лімфома кишечнику зустрічається в дорослих. У хворих спостерігаються виразки і перфорація слизової оболонки тонкої кишки.
При анапластичній крупноклітинній лімфомі поряд із пухлинами лімфатичних вузлів виявляють екстранодальні осередки ураження.
Лімфоми, клітини яких експресують антиген ЄМА+, мають більш агресивний перебіг. Хворіють як дорослі, так і діти. Лімфоми, клітини яких експресують антиген ЄМА—, зустрічаються тільки в дорослих і мають більш тривалий і ма- лосимптомний перебіг.
Клінічна класифікація НЗЛ була прийнята в 1973 р. В її основі лежить класифікація лімфогранулематозу (Ann-Arbor, 1971).
I стадія — локалізований пухлинний процес, уражуються лімфатичні вузли тільки однієї ділянки;
IE стадія — екстранодальне ураження та поширення процесу на суміжний орган;
II стадія — ураження лімфатичних вузлів кількох ділянок з одного боку діафрагми;
IIE стадія — поширення патологічного процесу на один із суміжних органів. Цю стадію також називають локально-регіональною;
III стадія — генералізований пухлинний процес, коли уражуються лімфатичні вузли кількох ділянок по обидва боки діафрагми;
IIIE стадія — приєднується ураження одного із суміжних органів;
IV стадія — дисемінований пухлинний процес, що характеризується ураженням печінки, кісток, шкіри та інших органів і систем;
V стадія — лейкемічна трансформація пухлинного процесу — лей- кемізація кісткового мозку і крові.
Крім того, у кожній стадії захворювання виділяють 2 фази захворювання: А — асимптомну, Б — з одним або кількома симптомами інтоксикації.
До симптомів інтоксикації відносять швидку втрату більше ніж 10 % маси тіла, температуру тіла понад 38 °С, нічне потіння.
Ураження кісткового мозку може спостерігатися при будь- якій морфологічній формі НЗЛ. При НЗЛ низького ступеня злоякісності (лімфоцитарній, що походить з малих лімфоцитів, центроцитарній, лімфоплазмоцитарній, волосатоклітинній) частота первинного ураження кісткового мозку становить від 86 до 99 %. У таких випадках картина крові і кісткового мозку нагадує таку при класичній формі хронічного лімфолейкозу.
При НЗЛ високого ступеня злоякісності (В- і Т-лімфо- бластній, центробластній, В-великоклітинній, імунобластній) ураження кісткового мозку спостерігається у 20—25 % випадків. Картина периферичної крові і кісткового мозку не відрізняється від такої при гострому лімфобластному лейкозі.
Диференціальну діагностику НЗЛ проводять із хронічним неспецифічним лімфаденітом, хворобою «від котячих подряпин», інфекційним мононуклеозом, туберкульозом лімфатичних вузлів. Діагноз встановлюють за допомогою біопсії лімфатичних вузлів, морфологічного дослідження відбитків пухлинної тканини та імунофенотипування.
Велике прогностичне значення має маса пухлини. Встановлено, що великі пухлини гірше піддаються лікуванню, ніж малі (навіть коли їх багато). Поганими прогностичними ознаками вважають симптоми загальної інтоксикації та важкий загальний стан хворого.
Лікування
Лікування призначають з урахуванням ступеня злоякісності лімфоми і стадії пухлинного процесу.
При НЗЛ проводять комплексну терапію. Застосовують поліхіміотерапію в комбінації з променевою терапією та хірургічними методами. Ефективність лікування оцінюють за такими критеріями:
повна ремісія — зникнення всіх проявів захворювання;
неповна ремісія — розміри уражених лімфатичних вузлів та екстранодальних інфільтратів зменшуються на 50 % і більше (за відсутності прогресії інших осередків);
клінічне поліпшення — зникнення клінічної симптоматики, розміри уражених лімфатичних вузлів зменшуються менше ніж на 50 %;
відсутність ефекту, прогресування захворювання.
Хірургічне лікування показане тільки при одиничних пухлинах органів травлення, щитоподібної і молочної залоз. Якщо виникають сумніви щодо радикальності проведеної операції, а також при лімфомах високого ступеня злоякісності після хірургічного втручання обов'язково проводять поліхіміотерапію.
Показання до видалення селезінки при НЗЛ такі: первинна лімфома селезінки без ознак генералізації, загроза розриву селезінки чи розвитку цитопенії, зумовленої гіперспленізмом (за наявності спленомегалії).
Променеву терапію як самостійний метод лікування при НЗЛ застосовують рідко. Як правило, призначають комбіновану терапію — «сандвіч» (2—3 цикли хіміотерапії, потім променева терапія та 2—3 цикли поліхіміотерапії).
Інтенсивність лікування та схема хіміотерапії залежать від гістологічного варіанту лімфоми, її імунофенотипу та від стадії захворювання.
У 1 стадії НЗЛ низького ступеня злоякісності до і після променевої терапії достатньо провести 2 цикли поліхіміотерапії. Потім знову призначають променеву терапію.
При бластній лімфомі високого ступеня злоякісності навіть у 1 стадії проводять хіміотерапію (по 3 курси до і після променевої терапії). Опромінюють тільки зони ураження. Профілактичне опромінення всіх груп лімфатичних вузлів недоцільне.
Комбіноване лікування ефективне в I-II стадіях НЗЛ. Повна ремісія протягом 5 років спостерігається більше ніж у 50 % хворих. У III-IV стадіях НЗЛ комбінована терапія не ефективніша за результатами за одну хіміотерапію, особливо в IV стадії, коли опромінення окремих ділянок має паліативний характер.
Кортикостероїдні гормони при НЗЛ застосовують досить рідко. Призначають преднізолон короткими курсами (5— 14 днів). Доза преднізолону — від 40—100 мг/м2 залежно від схеми поліхіміотерапії.
Поліхіміотерапію проводять короткими інтенсивними циклами з інтервалами 2—4 тиж.
Для встановлення чутливості пухлинної тканини до поліхіміотерапії необхідно провести не менше ніж 2 цикли лікування. І якщо вони були неефективними, то метод лікування слід замінити на інший.
Якщо вдалося досягти повної ремісії, проводять ще 2— З цикли консолідації ремісії, після чого лікування припиняють. У разі часткової ремісії лікування продовжують до повної ремісії або до рецидиву захворювання.
Слід зауважити, що в більшості хворих із НЗЛ низького ступеня злоякісності виявляють III-IV стадії захворювання. Оскільки такі лімфоми повільно прогресують, то тривалий час можна проводити монохіміотерапію циклофосфаном (по 200 мг на добу або по 400 мг через день). Можна призначити хлорбутин (лейкеран) — по 10 мг 5 разів на тиждень. Дозу хлорбутину в 1 -й тиждень можна збільшити до 20 мг на добу. Преднізолон у дозі 30—40 мг на добу посилює дію протипухлинних препаратів. Такі курси лікування повторюють через кожні 5—6 міс. Якщо монохіміотерапія не дає достатнього ефекту, проводять поліхіміотерапію за схемами COP, CVPP, LVPP, CHOP, CAMP тощо.
Схему COP можна посилити, призначивши протипухлинні антибіотики адріабластин (ACOP) чи блеоміцин (COP-Bleo) або обидва препарати (BACOP). Застосовують й інші хіміопрепарати: етопозид, теніпозид, флударабін (флудара), мітоксантрон, іфосфамід). Такі препарати зазвичай призначають при рецидивах захворювання та резистентних до лікування його формах.
НЗЛ високого ступеня злоякісності потребують проведення більш інтенсивної поліхіміотерапії. Погано піддаються лікуванню пухлини середостіння, для яких характерна швидка генералізація пухлинного процесу з ураженням легень, плеври, нирок і особливо кісткового мозку та ЦНС. Ураження кісткового мозку при бластних формах НЗЛ скорочують тривалість життя хворих унаслідок розвитку лейкемізації за типом гострого лейкозу. У таких випадках застосовують протоколи лікування хворих на ГЛЛ.
Прогноз при нелейкемічному НЗЛ визначають за міжнародним прогностичним індексом — IPI. Виділено 5 незалежних прогностичних показників: вік, стадія захворювання, загальний стан хворого (за шкалою ECOG), кількість зон екст- ранодального ураження, підвищення рівня лактатдегідрогенази в сироватці крові.
За сукупністю прогностичних ознак можна прогнозувати не тільки тривалість життя, але й результат лікування, ризик розвитку рецидивів, тривалість ремісій.
Важливе прогностичне значення мають гістологічний та імунофенотиповий варіант лімфоми, кількість зон пухлинного ураження, симптоми інтоксикації, розміри окремих пухлинних утворень, ураження середостіння і кісткового мозку, результати проведеного лікування.
Чим швидше настала ремісія, тим кращі віддалені результати лікування. Тому останніми роками тривалість лікування зменшують за рахунок його інтенсифікації.
Пухлини високого ступеня злоякісності швидко ростуть, тому вони потребують інтенсивної хіміотерапії.
Найчастіше застосовують циклофосфан і алкеран (мелфа- лан). Циклофосфан призначають у дозі 40—45 мг/кг, мелфа- лан — у дозі 1,5-1,7 мг/кг.
При ураженні ЦНС обов'язково використовують метотрексат, цитарабін (цитозар) і циклофосфан. Метотрексат призначають по 2—3 г/м2, цитарабін — по 3 г/м2. При ураженні мозкових оболон метотрексат уводять ендолюмбально по 12,5 мг/м2, а при поєднанні з цитозаром — по 5—30 мг/м2.
При первинній резистентності пухлин до лікування та рецидивах захворювання схему лікування змінюють. Після мо- нохіміотерапії проводять пол!хіміотерапію. Після лікування за схемами першої лінії використовують схеми другої лінії. Останні включають такі лікарські засоби, як антрацикліни, ето- позид, препарати нітрозосечовини, цитарабін.
Останніми роками для лікування окремих форм НЗЛ низького ступеня злоякісності застосовують а-інтерферони (лафе- рон, інтрон-А). Лікування а-інтерфероном проводять на фоні хіміотерапії, а також до і після неї. Застосування «-інтерферону в 1,5—2 рази збільшує кількість повних ремісій. Цей препарат застосовують у дозі 6—9 MO на добу на фоні поліхіміотерапії. Для підтримувальної терапії його призначають у дозі 3—5 MO на добу 3 рази на тиждень. Тривалість лікування — 12—18 міс.
Новим методом лікування НЗЛ є застосування MKAT проти антигену CD20. Призначають такі препарати, як мабтера або ритуксимаб. Монотерапія MKAT та лікування MKAT у поєднанні з хіміотерапією (за схемою CHOP) зумовлюють настання повної ремісії в 55 % хворих з НЗЛ помірного ступеня злоякісності. Ритуксимаб уводять внутрішньовенно в дозі 375 мг/м2 1 раз на тиждень. Курс лікування включає 4 таких інфузій.
Під час проведення поліхіміотерапії пригнічується кровотворення. Розвиваються нейтропенія та інфекційні ускладнення. Щоб запобігти таким ускладненням, застосовують такі препарати, як Г-КСФ і ГМ-КСФ.
Прогноз. Сучасні методи лікування дозволяють подовжити життя хворих, однак прогноз здебільшого несприятливий.
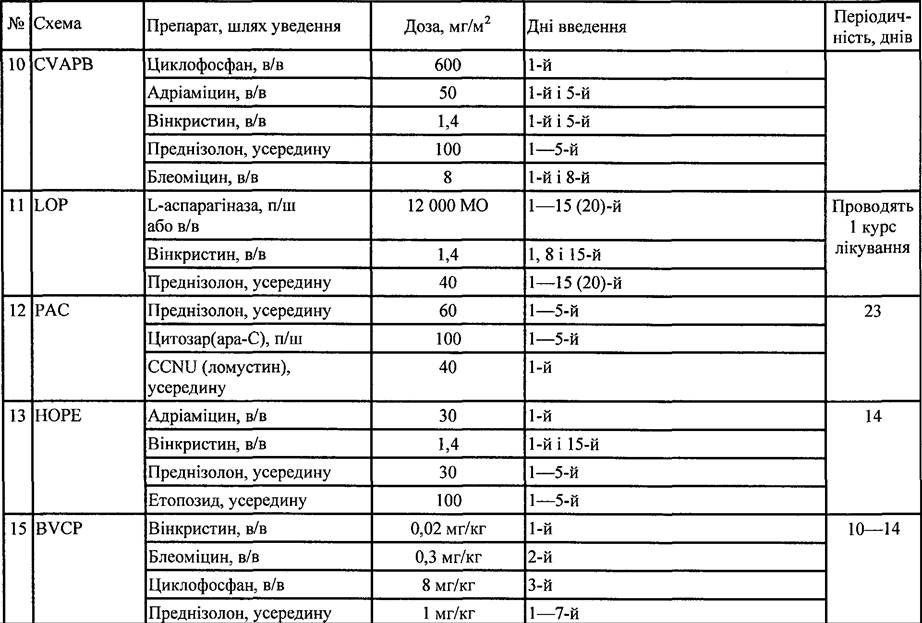
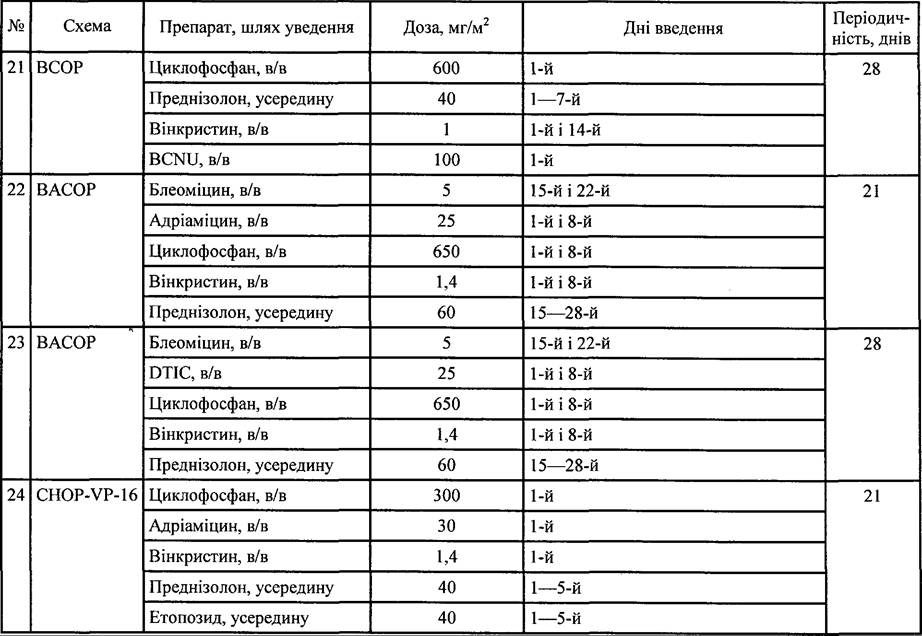
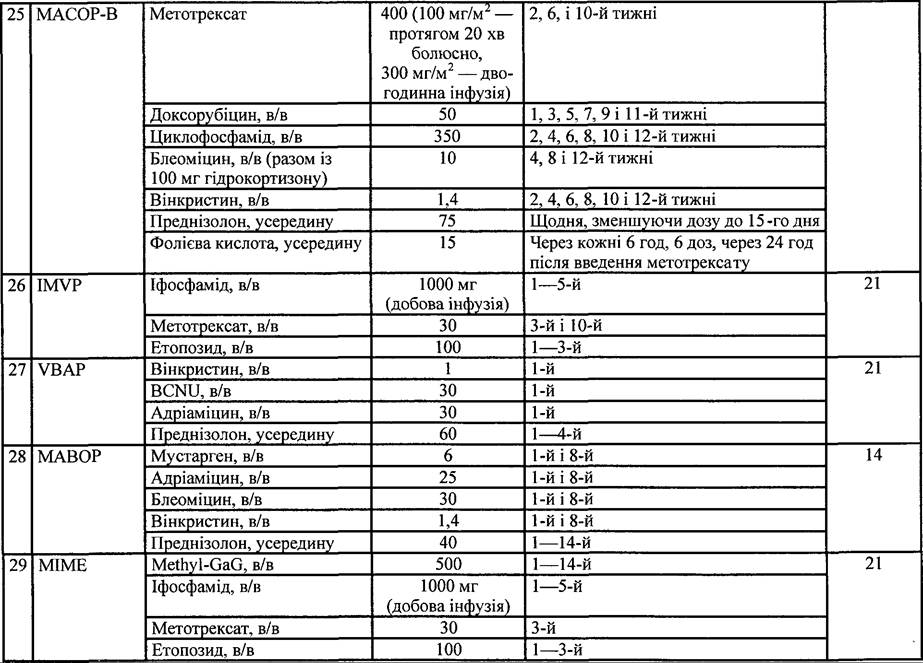
Таблиця 35. Схеми поліхіміотерапії, які застосовують при лікуванні НЗЛ
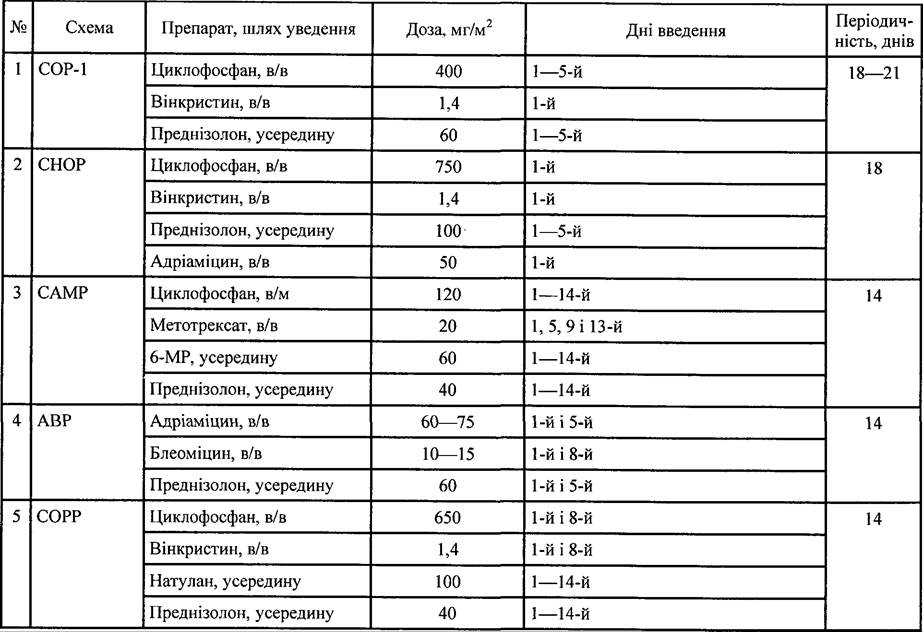
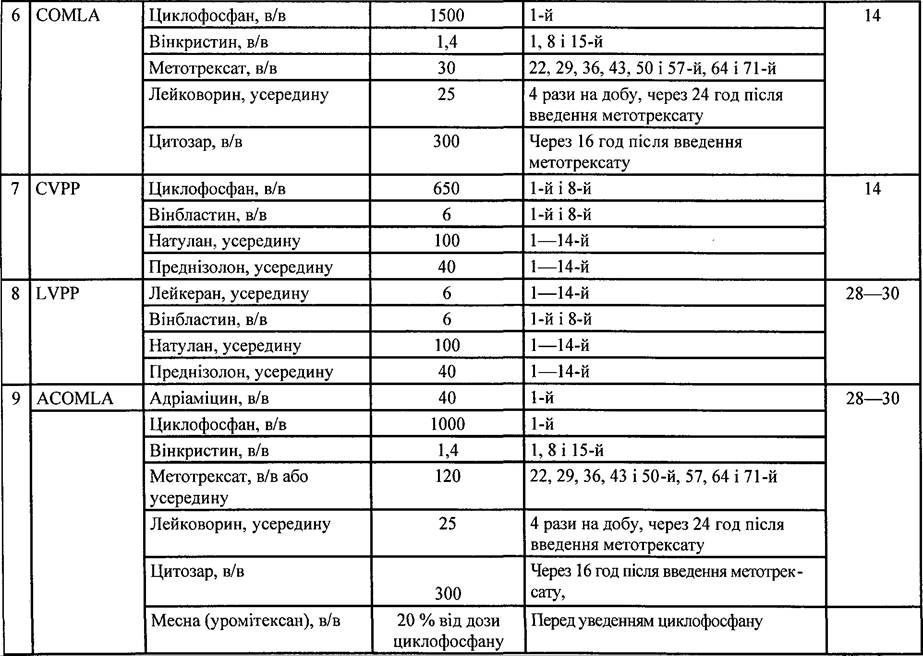
853

855
Еще по теме НЕХОДЖКІНСЬКІ ЗЛОЯКІСНІ ЛІМФОМИ:
- Злоякісні лімфоми
- 19.2. Негоджкінські лімфоми (лімфосаркоми)
- ЗЛОЯКІСНІ ПУХЛИНИ НОСА ТА ПРИНОСОВИХ ПАЗУХ
- 9.3.2. Злоякісні пухлини глотки
- 8.3.2. Злоякісні пухлини слинних залоз
- 8.4.2. Злоякісні пухлини порожнини носа та додаткових пазух
- ЗЛОЯКІСНІ ПУХЛИНИ ГЛОТКИ
- 8.1.2. Злоякісні пухлини губи
- 9.1.2. Злоякісні пухлини щитовидної залози
- ГЕМОБЛАСТОЗИ У ДІТЕЙ
- Розділ 19. Гемобластози
- 20.1. Пухлини нирок
- Розділ 17. Пухлини кісток
- Розділ 7. Пухлини шкіри
- Принципи класифікації пухлин
- Ріст пухлини.