5.1. Гипогонадизм
Гипогонадизм (тестикулярная недостаточность) – патологическое состояние, клиническая картина которого обусловлена стойким, часто необратимым снижением в организме уровня андрогенов (гипоандрогения) или ослаблением их действия (Козлов Г.
И., 2000). Гипогонадизм характеризуется недоразвитием половых органов, вторичных половых признаков и, как правило, бесплодием (Дедов И. И. [и др.], 2000).По характеру развития гипогонадизм (тестикулярная недостаточность) подразделяют на первичный и вторичный. Первичный гипогонадизм развивается как результат врожденных или приобретенных нарушений функции яичек. Его отличительным признаком является снижение циркуляции продуцируемых яичками гормонов (тестостерона и ингибина) при значительном возрастании уровня гонадотропинов (ЛГ и ФСГ) как результат реализации механизма отрицательной обратной связи. Поэтому первичный гипогонадизм называется гипергонадотропным. Наиболее часто первичный врожденный гипогонадизм имеет место при крипторхизме и генетических аномалиях, приводящих к нарушению развития и функции яичек. Первичные приобретенные нарушения тестикулярной функции могут быть индуцированы травмой яичек, действием на них неблагоприятных факторов среды (медикаментозных, химических, лучевых, температурных и др.), опухолью яичек и их поражением при инфекционных процессах.
Изолированное снижение секреции гонадотропинов или снижение продукции ФСГ и ЛГ в сочетании с выпадением других тропных функций гипофиза рассматривается как результат врожденного или приобретенного поражения гипофиза (вторичный гипогонадизм) или гипоталамуса (третичный гипогонадизм). Две последние формы характеризуются снижением секреции гонадотропинов, и их иногда объединяют под названием гипогонадотропный гипогонадизм.
При изолированной гонадотропной недостаточности основные клинические симптомы, как и при первичном гипогонадизме, обусловлены низким уровнем андрогенов (Козлов Г.
И., 2000). Вторичный гипогонадизм, как и первичный, подразделяется на врожденный и приобретенный. Наиболее частыми причинами вторичного врожденного гипогонадизма являются наследственные синдромы – опухоли, нарушения эмбрионального развития гипоталамуса и гипофиза. Вторичный приобретенный гипогонадизм развивается как проявление гипоили пангипопитуитаризма после инфекционно-воспалительных заболеваний, травм, опухолей гипоталамо-гипофизарной области.При скрытой недостаточности гипоталамо-гипофизарной системы может иметь место нормогонадотропный гипогонадизм, который характеризуется низкой продукцией андрогенов при нормальном уровне гонадотропных гормонов (Потемкин В. В., 1999). Нормогонадотропный гипогонадизм, возможно, связан с нарушением синтеза α-иβ-цепей гонадотропинов или с нарушением цирхорального ритма секреции ГнРГ.
Гипогонадизм может быть самостоятельным заболеванием (изолированный гипогонадизм) или одним из симптомов в структуре врожденных или приобретенных, в том числе эндокринных заболеваний (симптоматический гипогонадизм). Иногда у детей и подростков выявляют гиперпролактинемический гипогонадизм (Шабалов Н. П., 2003; Фролов Б. А., 2006).
При всех этих ситуациях пролактин, который в физиологических условиях стимулирует синтез тестостерона, секретируется в избытке и по короткой петле отрицательной обратной связи подавляет продукцию ЛГ и ФСГ, что и обусловливает развитие гипогонадизма. Кроме того, в генезе гиперпролактинемического гипогонадизма важная роль принадлежит нарушению конверсии тестостерона в периферических тканях в его активный метаболит 5a-дигидротестостерон, чем объясняется выраженность нарушений на фоне сравнительно небольшого снижения уровня тестостерона в крови.
Предполагается также, что гипогонадизм обусловлен центральным влиянием ПРЛ (возможно, через гипоталамус) на ГнРГ-секретирующие нейроны. Очевидно, что подобный сдвиг уровней ГнРГ приводит к ингибированию уровня аденогипофизарной секреции гонадотропинов, формируя их дефицит и, следовательно, ограничивая регуляторное влияние на яички.
Вместе с тем предполагается, что нарушение аденогипофизарной секреции ЛГ и ФСГ может быть связано со стимуляцией ПРЛ секреции кортикотропина и надпочечниковых андрогенов, обусловливающих в отношении гонадотропинов включение «петли» отрицательной обратной связи. При этом действие такой «петли» в значительной степени затрагивает секрецию ЛГ. Кроме того, допускается прямое ингибирующее воздействие высоких концентраций ПРЛ на рецепцию ГнРГ в аденогипофизе и гонадотропных гормонов в яичках.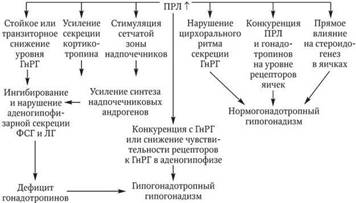
Рис. 5.1. Участие пролактина в формировании различных вариантов гипогонадизма у мальчиков и подростков
Таким образом, можно сделать вывод, что гиперпролактинемический гипогонадизм как отдельно взятую форму выделять не совсем правомочно (рис. 5.1).
Повышение пролактина по одним механизмам приводит к снижению гонадотропинов, ГнРГ и формированию гипогонадотропного гипогонадизма (вторичного или третичного), а по другим – уровни указанных гормонов находятся в пределах нормы, но формируется нормогонадотропный гипогонадизм. Эта разновидность гипогонадизма прежде всего связана с воздействием высоких уровней ПРЛ на цирхоральный ритм секреции ГнРГ и его нарушение и, как следствие, происходит снижение выработки гонадотропинов. Кроме того, ПРЛ может взаимодействовать непосредственно с рецепторами гонадотропинов на яичках и подавлять стероидогенез (снижать выработку тестостерона).
Гиперпролактинемия может проявляться как самостоятельное заболевание (микро– и макропролактинома, идиопатическая гиперпролактинемия) в сочетании с другой гипоталамо-гипофизарной патологией (соматопролактиномы, кортикотропиномы, синдром «пустого» турецкого седла, внутричерепная гипертензия, лимфоцитарный гипофизит и др.), как вторичный патологический процесс при первичном гипотиреозе, печеночной и почечной недостаточности, нейрогенных нарушениях, а также при применении наркотиков и медикаментов, обладающих антидофаминергической активностью (Дедов И.
И. [и др.], 2000).Клиника разных форм гипогонадизма достаточно однообразна и зависит от возраста пациента и времени начала заболевания. Объем яичек у мальчиков уменьшен, они могут быть расположены в мошонке, но чаще первым симптомом врожденного гипогонадизма является внемошоночное их расположение (одно– или двусторонний крипторхизм). Иногда яички аплазированы, часто имеют плотную или дряблую консистенцию, в пубертатном возрасте их увеличения не происходит. Половой член уменьшен в размерах, мошонка без складок, подтянута, вторичные половые признаки в пубертатном возрасте отсутствуют или выражены слабо.
Недостаточность андрогенов приводит к постепенному формированию экстрагенитальных симптомов гипогонадизма: евнухоидизма, ожирения, уменьшения костной и мышечной массы, нередко высокорослости. Евнухоидные пропорции тела характеризуются удлинением конечностей при относительно коротком туловище и преобладании ширины таза над шириной плеч. Ожирение может возникнуть еще в допубертатном возрасте, чаще в легкой степени, поскольку увеличение жировой ткани сочетается со слабым развитием мышц и уменьшением костной массы. Типично перераспределение жировой ткани по женскому типу с преимущественным отложением ее в область бедер, живота, груди (ложная гинекомастия), иногда встречается истинная гинекомастия. Как правило, у мальчиков, в связи с задержкой формирования костной ткани и отставанием дифференцировки скелета, рост продолжается дольше обычного, но окончательный рост может быть выраженный. Мышцы слабые, дряблые. Экстрагенитальные признаки гипогонадизма обычно начинают формироваться в 9 – 12 лет и становятся очевидными у подростков старше 13 – 14 лет, редко в более раннем возрасте.
Симптоматический гипогонадизм чаще диагностируется в раннем возрасте. Основанием для диагностики заболевания обычно является сочетание ряда фенотипических признаков, пороков развития и симптомов, не обусловленных гипогонадизмом.
Наиболее частые симптомы гипогонадизма у мальчиков представлены в табл.
5.1.Классификация основных форм гипогонадизма, предложенная профессором Н. П. Шабаловым, представлена в табл. 5.2.
Классификация гипогонадизма у мальчиков
I. ГИПЕРГОНАДОТРОПНЫЙ (ПЕРВИЧНЫЙ) ГИПОГОНАДИЗМ
А. Врожденный.
1. Нарушение развития гонад:
а) дисгенез семенных канальцев – синдром Клайнфелтера и его варианты:
• синдром Клайнфелтера (классический);
Таблица 5.1
Наиболее частые симптомы гипогонадизма у мальчиков

Таблица 5.2
Классификация основных форм гипогонадизма у мальчиков (Шабалов Н. П., 2003)



• синдром Клайнфелтера 46XY (ложный);
• синдром с 46ХХ;
б) микроделеции Y-хромосомы (AZFb, AZFc-участков);
в) первичная анаплазия тестикул (синдром дель Кастильо или синдром одних клеток Сертоли) – делеция AZFa-участка Y-хромосомы;
г) анорхизм;
д) крипторхизм;
е) синдром Ульриха – Нунан;
ж) синдром дисгенезии тестикул (мужской вариант синдрома Шерешевского – Тернера);
з) синдром истинного агонадизма;
и) синдром рудиментарных яичек;
й) дисгенез гонад или синдром Свайера;
к) агенез клеток Лейдига;
л) синдром 46ХХ у мужчин и подростков или синдром де ля Шапеля;
м) синдром двух или трех Y (47XYY, 48XYYY);
н) синдром неподвижности ресничек.
2. Нарушение развития протоков:
а) аплазия вольфовых протоков;
б) дисгенез вольфовых протоков (кистозный фиброз);
в) синдром персистенции мюллеровых протоков (hernia uteri inguinale).
3. Нарушение дифференцировки урогенитального синуса и половых органов (мужской псевдогермафродитизм):
а) недостаточность биосинтеза андрогенов:
• недостаточность 20, 22-десмолазы;
• недостаточность 3â-гидроксистероидной дегидрогеназы;
• недостаточность 17a-гидроксилазы;
• недостаточность 17,20-десмолазы;
• недостаточность 17â-гидроксистероидной дегидрогеназы;
б) недостаточность биологического действия андрогенов:
• недостаточность рецепторов к андрогенам;
• синдром полной резистентности к андрогенам – синдром тестикулярной феминизации или синдром Морриса;
• синдром неполной резистентности к андрогенам и синдром Рейфенштейна;
• микропенис (синдром микрогенитализма);
• недостаточность 5a-редуктазы типа 2.
Б. Приобретенный.
1. Инфекционные агенты (орхит после паротита, лепра и др.).
2. Травма яичек.
3. Радиоактивное облучение яичек.
4. Аутоиммунная недостаточность яичек.
5. Идиопатическая олигозооспермия и азооспермия.
II. ГИПОГОНАДОТРОПНЫЙ (ВТОРИЧНЫЙ И ТРЕТИЧНЫЙ) ГИПОГОНАДИЗМ
А. Врожденный.
1. Идиопатический гипогонадизм, сформированный в результате вредных воздействий в антенатальном, интранатальном и постнатальном периоде.
2. Ольфактогенитальный синдром (синдром Каллмана).
3. Изолированная недостаточность ЛГ (синдром Маккола или синдром «фертильного евнуха», синдром Паскуалини).
4. Преимущественный или иногда изолированный дефицит ФСГ.
5. Адипозогенитальная дистрофия (синдром Бабинского – Фрелиха).
6. Гипогонадизм со сниженной секрецией ЛГ, ФСГ и кортикотропина (синдром Медока).
7. Синдром неполной маскулинизации со снижением ЛГ, ФСГ и Т.
8. Врожденная гипоплазия надпочечников.
9. Гипогонадотропная недостаточность при генетических синдромах.
10. Гемохроматоз.
11. Изолированное повышение ФСГ в крови.
12. Гипогонадизм, связанный с тотальной недостаточностью гипофиза при гипофизарном нанизме.
Б. Приобретенный.
1. Приобретенная недостаточность гонадотропинов – гипопитуитаризм (воспалительные процессы, травмы с переломом основания черепа, сосудистые аневризмы).
2. Патологические состояния, связанные с повышением секреции ПРЛ.
3. Гипогонадизм, связанный с тотальной недостаточностью гипофиза при гипофизэктомии.
4. Ятрогенный и лекарственный гипогонадизм.
III. НОРМОГОНАДОТРОПНЫЙ (ВРОЖДЕННЫЙ И ПРИОБРЕТЕННЫЙ) ГИПОГОНАДИЗМ
Патологические состояния, связанные с повышением секреции ПРЛ.
IV. КОРРЕЛЯТИВНЫЙ ГИПОГОНАДИЗМ
Формируется на фоне системных заболеваний (декомпенсированного сахарного диабета, цирроза печени и др.).
Классификация патологических состояний, связанных с повышением секреции ПРЛ (Балаболкин М. И. [и др.], 2002)
1. Заболевания, приводящие к нарушению функции гипоталамуса:
• инфекции (менингит, энцефалит и др.);
• гранулематозные и инфильтративные процессы (саркоидоз, гистиоцитоз, туберкулез и др.);
• опухоли (глиома, менингиома, краниофарингиома, герминома и др.);
• травмы (разрыв ножки мозга, кровоизлияние в гипоталамус, блокада портальных сосудов;
• метаболические нарушения (цирроз печени, хроническая почечная недостаточность).
2. Поражение гипофиза:
• пролактинома (микро– или макроаденома);
• идиопатическая форма гиперпролактинемии;
• смешанная соматотропно-пролактиновая аденома;
• другие опухоли (соматотропинома, кортикотропинома, тиротропинома, гонадотропинома);
• синдром «пустого» турецкого седла;
• краниофарингиома;
• гормонально-неактивная, или «немая», аденома;
• аденомы гипофиза, сочетающиеся с гипопитуитаризмом;
• интраселлярная герминома, менингиома, киста или киста кармана Ратке.
3. Другие заболевания:
• первичный гипотиреоз;
• эктопированная секреция гормонов;
• врожденная гиперплазия коры надпочечников;
• хроническая почечная недостаточность;
• цирроз печени;
• повреждения грудной клетки (Herpes zoster, перелом ребер и другие травмы).
4. Лекарственные препараты:
• блокаторы дофамина (сульпирид, метоклопрамид, домперидон, нейролептики, фенотиазиды);
• антидепрессанты (имипрамин, амитриптиллин, галоперидол);
• блокаторы кальциевых каналов (верапамил);
• адренергические ингибиторы (резерпин, альфа-метилдофа, альдомет, карбидофа, бензеразид);
• эстрогены;
• блокаторы Н2-рецепторов (циметидин);
• опиаты и кокаин;
• тиролиберин, ВИП.
Краткая характеристика основных форм гипогонадизма у мальчиков и подростков
ПЕРВИЧНЫЙ (ГИПЕРГОНАДОТРОПНЫЙ) ГИПОГОНАДИЗМ
Синдром Клайнфелтера (дисгенезия семенных канальцев)
Заболевание, обусловленное аномалией половых хромосом, характерным симптомом которого является нарушение сперматогенеза. Частота у лиц с мужским фенотипом 1: 500 – 1: 700, среди пациентов с бесплодием – 1:9.Надолю синдрома приходится более половины всех хромосомных аномалий половой системы.
Этиология: кариотип 47XXY, 48XXXY, 49XXXXY (синдром Жозефа) или мозаицизм.
Патогенез связан с хромосомными нарушениями: присутствие в клетках (чаще из-за нерасхождения в мейозе) дополнительной Х-хромосомы. Кариотип 47XXY. Возможны кариотипы: 48XXXY, 48XXYY. Мозаичный кариотип наиболее часто представлен 46XY/47XXY:
• в эмбриональном периоде – нормальное формирование яичек и мужских половых органов (обусловлено присутствием в кариотипе Y-хромосомы);
• в пубертатном периоде – дегенеративные изменения яичек (гиперплазия клеток Лейдига и фиброз, гиалиноз семенных канальцев) с нарушением их нормального развития и функции;
• недостаточная секреция яичками Т, приводящая к значительному повышению уровня ЛГ и ФСГ и формированию патологических проявлений.
Клинические проявления:
• клинические признаки появляются в постпубертатном, реже – в пубертатном периоде, вторичные половые признаки могут быть нормальными, слаборазвитыми или отсутствовать;
• в допубертатном возрасте обращает внимание ускоренный рост и нарушение поведения; иногда при рождении имеется крипторхизм;
• в пубертатном возрасте наиболее типичный признак – уменьшенные и уплотненные яички, типичны высокорослость, ожирение, гинекомастия и евнухоидизм, слабое развитие мускулатуры, психическая вялость, эмоциональная неустойчивость;
• в большинстве случаев развитие полового члена и мошонки нормальное с пубертатными изменениями, однако возможен микрогенитализм;
• встречается правильное телосложение с хорошо развитыми вторичными половыми признаками, нормальными размерами полового члена при сохранности копулятивной функции. В таких случаях единственный симптом – бесплодие из-за азооспермии (преимущественно у взрослых), падение либидо и потенции – после 30 – 35 лет;
• тельца Бара (включения в ядерной мембране, характерные только для ХХ-клеток) в букальном мазке. Линия 47XXY-клеток или множественные линии клеток с мозаичными вариантами в культуре лейкоцитов.
Анорхизм
Эмбриональная аномалия, характеризующаяся отсутствием яичек у гено– и фенотипических мальчиков.
Этиология: внутриутробное, возможно генетически детерминированное, поражение яичек.
Патогенез: гибель яичек происходит на 20-й неделе эмбрионального развития, что исключает их дальнейшее влияние на морфогенез полового тракта. В пубертатном периоде не происходит развития вторичных половых признаков в связи с дефицитом Т. Яички в мошонке, паховых каналах и брюшной полости отсутствуют; иногда отсутствует и сама мошонка.
Клинические проявления:
• яички в мошонке и паховых каналах отсутствуют с рождения;
• при рождении наружные половые органы сформированы правильно, обычных размеров, но иногда их размеры уменьшены;
• признаки полового созревания в пубертатном возрасте отсутствуют, но иногда появляется скудное оволосение в андрогензависимых зонах;
• в допубертатном возрасте рост средний, телосложение маскулинное. В допубертатном и пубертатном возрасте появляется ожирение, ускорение роста, формируется евнухоидный тип телосложения. Интеллект сохранен;
• значительное ослабление либидо, отсутствие, как правило, эрекции и поллюций;
• половая ориентация чаще по мужскому типу, но крайне неустойчива;
• отсутствие (в отличие от крипторхизма) реакции Т плазмы на ХГЧ (1500 ЕД/сут на протяжении 5 дней).
Крипторхизм
Задержка яичка в процессе его опускания во внутриутробном периоде (на 7-м месяце) из брюшной полости в мошонку. Чаще (50,8 %) правосторонний, двусторонний – в 13,9 % случаев.
Этиология:
• механические (анатомические) препятствия в паховых каналах, укорочение и недоразвитие семенного канатика и его сосудов, отсутствие направляющей связки и ее внутрибрюшинные сращения;
• гормональная (дефицит ЛГ и ФСГ);
• снижение чувствительности яичек и других тканей к гормонам.
Патогенез:
• дегенеративные изменения в неопустившихся в мошонку яичках: более высокая температура в паховом канале и брюшной полости (на 2 – 6 °C), чем в мошонке, что активирует аутокаталитические процессы в тестикулярной ткани (при участии расщепляющей лейцинамид пептидазы), травматизация яичка окружающими тканями, аутоиммунные механизмы в связи с нарушением гематотестикулярного барьера;
• степень поражения яичка тем больше, чем выше оно расположено от мошоночной полости и чем длительнее его дистопическое положение;
• нарушения эндокринной и сперматогенной функций яичка, способные привести к бесплодию.
Клинические проявления:
• отсутствие яичек (яичка) в мошонке;
• недоразвитие соответствующей половины мошонки при одностороннем крипторхизме;
• возможное недоразвитие половых органов и вторичных половых признаков при двустороннем крипторхизме;
• отклонение от нормального развития ЦНС: недостаточность психомоторики, речевые затруднения и др.;
• другие симптомы как результат нарушения внутриутробного развития (узкое нёбо, врожденный экзофтальм, диспластичные ушные раковины, паховая и пупочная грыжи);
• осложнения крипторхизма (гипогенитализм, бесплодие, ущемление и заворот неопустившегося яичка, его малигнизация злокачественное перерождение).
Сертоли-клеточный синдром (синдром дель Кастильо)
Первичная герминативная аплазия яичек.
Этиология окончательно не изучена. Имеются данные о повреждении AZFa-участка длинного плеча (Yq11) Y-хромосомы, отвечающего за выработку зооспермического фактора. Выявляется связь синдрома с врожденным отсутствием зародышевых клеток.
Патогенез:
• повреждаются только герминативные клетки яичка при нормально сформированной гонаде;
• в результате возникает атрофия семенного эпителия, клетки Сертоли не повреждаются;
• аналогичные изменения наблюдаются при радиоактивном облучении, тяжелых заболеваниях нервной системы (рассеянный склероз, травмы черепа и позвоночника).
Клинические проявления:
• мужской кариотип (46XY) при нормальном половом и физическом развитии с сохранением либидо, спонтанной и адекватной эрекции. Поэтому заболевание в детском возрасте не диагностируют из-за отсутствия отклонений в половом и физическом развитии;
• характерно уменьшение размеров яичек при нормальном развитии наружных половых органов и вторичных половых признаков, бесплодие (в эякуляте – азооспермия), крипторхизм встречается редко;
• при гистологическом исследовании яичек наблюдается уменьшение в диаметре семенных канальцев, полное отсутствие сперматогенного эпителия при наличии вакуолизированных клеток Сертоли и скоплении клеток Лейдига в интерстиции;
• нормальный уровеньТиЛГпризначительном повышении ФСГ.
Первичная недостаточность (агенез) клеток Лейдига
Этиология: кариотип 46XY. Генетически обусловленные дефекты биосинтеза Т.
Патогенез связан с недостаточным влиянием Т в периоде полового созревания.
Клинические проявления:
• яички уменьшены, плотноватой консистенции, возможен крипторхизм;
• при рождении наружные гениталии сформированы правильно, отсутствует их пубертатное увеличение;
• клиника евнухоидизма с ожирением, недоразвитием половых органов и отсутствием полового оволосения формируется в пубертатном возрасте;
• снижение Т, увеличение ЛГ, ФСГ в норме, отсутствие реакции Т на 1– и 3-дневное введение ХГЧ.
Синдром Ульриха – Нунан (синдром Тернера)
Частота1:16000лицмужского пола.
Этиология: кариотип 46XY. Аутосомно-доминантный тип наследования.
Патогенез связан с нарушением половой дифференцировки.
Клинические проявления:
• у части мальчиков может быть в виде крипторхизма, микропениса и гипоплазии мошонки;
• иногда в виде евнухоидизма, формирующегося в пубертатном возрасте;
• крыловидные складки на шее, «треугольное» лицо, вальгусная деформация локтевых суставов, низкорослость, лимфатические отеки кистей и стоп, птоз, бочкообразная грудная клетка, деформация или низкое расположение ушных раковин, гипертелоризм, антимонголоидный разрез глаз, микрогнатия, высокое готическое нёбо, низкая линия волосяного покрова головы, наличие пороков сердца или крупных сосудов (стеноз легочной артерии), умственная отсталость;
• недостаточно выраженные вторичные половые признаки;
• гипотрофированные маленькие яички, сперматогенез отсутствует или наблюдается олигозооспермия различной степени.
Синдром 46ХХ у мужчин, или синдром де ля Шапеля
Этиология: генотип 46XX, фенотип мужской. Предполагается транслокация очень малой части Y-хромосомы либо на Х-хромосому, либо на аутосому. Допускается, что интактная Y-хромосома сохранилась в одной из линий стволовых клеток при мозаицизме.
Патогенез связан с нарушением половой дифференцировки.
Клинические проявления:
• такие же, как у пациентов с мозаицизмом XXY/XY;
• недоразвитие яичек.
Синдром неподвижности ресничек
Этиология: дефект гена динина на хромосоме 1р35.1.
Патогенез: выявлен дефицит белковой карбоксилметилазы – фермента, необходимого для подвижности сперматозоидов и подвижности ресничек на реснитчатом эпителии в других органах.
Клинические проявления:
• нарушение подвижности сперматозоидов вплоть до ее отсутствия, часто в сочетании с нарушением функции реснитчатого эпителия легочного тракта и соответственно с возникновением бронхитов и бронхоэктазов. Иногда наблюдается изолированное нарушение подвижности сперматозоидов;
• бесплодие при нормально развитых внутренних и наружных половых органах.
Синдром полной резистентности к андрогенам – синдром тестикулярной феминизации или синдром Морриса
Этиология: кариотип 46XY. Точечные мутации гена рецептора к андрогенам (чаще в 5-м и 7-м экзонах) на длинном плече Х-хромосомы (Хq11 – 12).
Патогенез:
• нарушение воздействия тестостерона (дигидротестостерона) на рецепторы ядер, клеток тканей-мишеней, к которым в первую очередь относятся ткани урогенитального синуса;
• в период эмбрионального развития первичная гонада трансформируется в тестикулы, продуцирующие Т. Нормальная функция клеток Сертоли и своевременное, достаточное количество антимюллерового фактора способствуют полному регрессу протоков Мюллера;
• наличие резистентности к андрогенам в клетках-мишенях является причиной трансформации урогенитального синуса в наружные половые органы по женскому типу (влияние эстрогенов коры надпочечников эмбриона и сохранение чувствительности к эстрогенам клеток урогенитального синуса).
Клинические проявления:
• женский фенотип;
• первичная аменорея;
• отсутствие или скудное проявление вторичных половых признаков (оволосение в подмышечных впадинах и на лобке);
• наружные половые органы по женскому типу, клитор обычных для женщин размеров, влагалище короткое и слепо оканчивающееся, внутренние женские половые органы отсутствуют;
• яички располагаются у внутреннего или наружного отверстия или по ходу пахового канала, изредка в области больших половых губ (определяются пальпаторно или на УЗИ);
• сохраняется либидо, способны к половой жизни, однако бесплодны;
• Т в норме или повышен, отсутствует связывание андрогенов в фибробластах кожи, взятых из области наружных половых органов.
Синдром неполной резистентности к андрогенам и синдром Рейфенштейна (синдром Gilbert – Dreyfus, или синдром Lubs – Rosewater)
Наследственное сцепленное с Х-хромосомой рецессивное заболевание.
Этиология: кариотип 46XY. Точечные мутации гена (374 варианта) на Х-хромосоме, ответственного за синтез рецептора к андрогенам.
Патогенез: сходный с патогенезом при синдроме Морриса.
Клинические проявления:
• гипоспадия, сочетающаяся с недоразвитием полового члена, искривленного книзу с короткой уздечкой, гинекомастия, евнухоидизм;
• крипторхизм, атрофия семенных канальцев, нередко азооспермия;
• мошонка развита нормально, яички слегка гипоплазированы;
• много общих черт с синдромом Клайнфелтера, однако кариотип 46XY;
• оволосение на лице и в подмышечных впадинах скудное, на лобке – по женскому типу;
• увеличение содержанияТиЛГвкрови;
• при гистологическом исследовании биоптата яичек обнаруживается гиалиноз и фиброз семенных канальцев (поражение в постпубертатном периоде); клетки Лейдига – в достаточном количестве.
Двусторонний орхит вследствие эпидемического паротита
Этиология: РНК-содержащий вирус эпидемического паротита из рода парамиксовирусов.
Патогенез:
• гематогенное распространение вируса из области входных ворот (слизистая рта, дыхательных путей, конъюнктивы) с возможным проникновением его в ЦНС и различные железистые органы, включая яички (в 20 – 30 % случаев эпидемического паротита);
• развитие, как правило, двустороннего воспалительного процесса с очаговой, а впоследствии диффузной инфильтрацией интерстиция;
• при поражении клеток Лейдига возможно развитие гипогонадизма;
• характерна обратимость процесса и редкое развитие бесплодия, имеющее место при прямом повреждении семенных канальцев.
Клинические проявления:
• выраженный болевой синдром в области яичек;
• гипертрофия яичек;
• лихорадка.
Опухоли яичка
Встречаются в любом возрасте, но преимущественно от 20 до 40 лет. Обычно односторонние. Доброкачественные опухоли составляют лишь 0,8 % всех новообразований яичка. Злокачественные опухоли яичка составляют 1 – 2 % всех злокачественных новообразований у мужчин.
Этиология: канцерогенные факторы (физические, химические, биологические), вызывающие мутагенный эффект.
Патогенез:
• ведущий фактор патогенеза – нарушение гормональных взаимодействий между гипофизом и яичками. Снижение функции яичек индуцирует гиперсекрецию гипофизарных гонадотропинов, что приводит к гиперстимуляции атипичных клеток;
• при крипторхизме опухоль в неопустившемся яичке развивается в 65 раз чаще, чем в яичке, находящемся в мошонке.
Клинические проявления:
• наиболее типичны болезненные при пальпации образования в мошонке;
• при гормонально-активной опухоли из клеток Лейдига (лейдигома) характерна гиперпродукция андрогенов, приводящая к преждевременному половому развитию;
• при гормонально-активной опухоли из клеток Сертоли (сертолиома, хорионэпителиома) наблюдается билатеральная гинекомастия, феминизация;
• маркеры опухолей яичка – a-фетопротеин, хорионический гонадотропин и лактатдегидрогеназа. При гормонально-активных опухолях имеют место высокие уровни андрогенов и (или) эстрогенов.
Другие формы первичного приобретенного гипогонадизма
Этиология: травмы яичек, в том числе родовые, при ягодичном предлежании плода, хирургические вмешательства, перекрут семенного канатика, орхит.
Патогенез:
• гибель (или резкая гипоплазия) яичек вследствие их механического повреждения, а также в результате воспалительного процесса;
• аутоагрессия (наличие аутоантител к тестикулярной ткани) у некоторых мальчиков-подростков;
• нарушения эндокринной и сперматогенной функций яичек.
Клинические проявления:
• в допубертатном периоде происходит формирование типичного евнухоидного синдрома и отставание в размерах полового члена с нарушением развития кавернозных тел;
• после 14 лет на фоне евнухоидных пропорций тела наблюдается гипогенитализм с отсутствием вторичных половых признаков (либидо, поллюций и адекватных эрекций);
• значительное отставание «костного» возраста от фактического.
• остеопороз.
ВТОРИЧНЫЙ И ТРЕТИЧНЫЙ ГИПОГОНАДИЗМ
Синдром Каллмана (ольфакто-генитальная дисплазия) I, II и III типов
Комплекс наследственных аномалий, характеризующихся сочетанием гипогонадизма с аносмией или гипосмией. По данным некоторых авторов (Козлов Г. И., 2000), частота синдрома Каллмана не уступает частоте синдрома Клайнфелтера.
Этиология: I тип обусловлен мутациями в гене KALIGI (Xp22.3). Тип наследования – аутосомно-рецессивный. При II типе отмечается генетическая гетерогенность, при III типе – Х-сцепленный рецессивный и аутосомно-доминантный типы наследования. При всех вариантах нарушена гонадолиберинсекретирующая функция нейронов, а также функция нейронов обонятельных луковиц. Дефекта гена ГРГ нет.
Патогенез:
• врожденный дефект развития гипоталамуса, приводящий к патологии обонятельных анализаторов, а также к снижению секреции ГнРГ;
• как результат дефицита ГнРГ – снижение продукции ЛГ и ФСГ гипофизом;
• формирование вторичного гипогонадизма с нарушением обоняния.
Клинические проявления:
• евнухоидизм, нередко сопровождающийся одно– или двусторонним крипторхизмом с паховой дистопией гипоплазированных яичек. В ряде случаев имеется только их гипоплазия;
• при рождении выявляют микропенис и недоразвитие мошонки; в пубертатном возрасте отсутствует их увеличение;
• снижение чувствительности к запахам (аносмия или гипосмия) в результате агенезии обонятельных центров мозга – при всех вариантах синдрома;
• при I типе бывает билатеральная синкинезия, атаксия, агенезия почки. При II типе – умственная отсталость, атрезия хоан, нейросенсорная глухота, пороки сердца, низкий рост. При III типе – расщепление губы и нёба, гипотелоризм, агенезия почки;
• при рентгенологическом исследовании костей наблюдается запаздывание возрастной дифференцировки.
Изолированная недостаточность ЛГ или синдром Паскуалини (синдром «фертильных евнухов»)
Этиология, как правило, связана с имеющейся недостаточностью ЛГ гипофиза вследствие мутации в гене бета-субъединицы ЛГ.
Патогенез:
• врожденная изолированная недостаточность секреции ЛГ;
• снижение продукции Т как следствие дефицита ЛГ, необходимого для поддержания нормального уровня ФСГ в плазме, обеспечивающего сохранность всех стадий сперматогенеза;
• механизм формирования олигозооспермии неясен.
Клинические проявления:
• при рождении выявляют микропенис и недоразвитие мошонки, в пубертатном возрасте отсутствует их развитие, в препубертатном возрасте типично ожирение, в дальнейшем – евнухоидизм, отсутствие полового созревания;
• чаще всего выявляют одно– или двусторонний крипторхизм с паховой дистопией гипоплазированных яичек;
• в ряде случаев имеется только уменьшение размеров яичек, однако они могут быть и нормальными;
• скудное оволосение лобка, подмышечных впадин, лица;
• нарушение половой функции, бесплодие;
• малый объем эякулята, низкая подвижность сперматозоидов, олигозооспермия, уменьшение содержания фруктозы в семенной жидкости;
• возможна (редко) гинекомастия;
• чаще высокий рост, интеллект нормальный.
Синдром Мэддока
Редкая форма вторичного гипогонадизма в сочетании с гипокортицизмом.
Этиология: мутация гена DAX1, контролирующего эмбриональное развитие аркуатных ядер гипоталамуса и надпочечников.
Патогенез:
• дефицит гонадотропных гормонов гипофиза и АКТГ;
• гипогонадизм в сочетании с вторичным хроническим гипокортицизмом.
Клинические проявления:
• врожденный двусторонний крипторхизм и микроорхидизм.
У некоторых пациентов крипторхизм отсутствует;
• евнухоидизм в сочетании с симптомами вторичной хронической надпочечниковой недостаточности (общая слабость, потеря массы тела, гипотония);
• половой член и мошонка гипоплазированы, отсутствуют пубертатные изменения;
• симптомы недостаточности надпочечников;
• признаки преждевременного старения.
Адипозогенитальная дистрофия (синдром Бабинского – Фрелиха)
Этиология: инфекционное, травматическое, хирургическое или лучевое поражение ЦНС. Чаще опухоль гипоталамо-гипофизарных отделов мозга, гистиоцитоз Х. может развиваться с наличием гемохроматоза и выраженной талассемией.
Патогенез: формирование симптоматики за счет нарушения функционирования гипоталамо-гипофизарных отделов ЦНС.
Клинические проявления:
• яички при рождении в мошонке, у подростков – гипотрофированы, возможна псевдоретенция;
• у подростков нет пубертатных изменений, отсутствует эрекция, исчезает складчатость и пигментация мошонки;
• вторичные половые признаки отсутствуют или регрессируют;
• гипогонадизм, как правило, сочетается с ожирением, снижением скорости роста, иногда гипотиреозом, несахарным диабетом, нарушением зрения, неврологической симптоматикой в различных комбинациях.
Синдром Prader – Willi
Этиология: у большинства обнаруживают делецию 15q11.2 – q13 отцовской хромосомы или дисомию того же фрагмента 15-й хромосомы материнского происхождения. Частота – 1:25000.
Патогенез не изучен.
Клинические проявления:
• двусторонний крипторхизм и гипоплазия яичек с рождения;
• половой член и мошонка резко гипоплазированы, отсутствуют их пубертатные изменения;
• вторичные половые признаки отсутствуют;
• в раннем возрасте имеется мышечная гипотония вплоть до атонии, снижение рефлексов. С 2 – 3 месяцев появляется полифагия, высокий болевой порог, олигофрения. С 1 – 1,5 лет развивается диэнцефальное ожирение, а в подростковом возрасте – сахарный диабет.
Синдром Laurence – Moon
Этиология: заболевание с аутосомно-рецессивным типом наследования.
Патогенез: не изучен.
Клинические проявления:
• двусторонний крипторхизм и микроорхидизм с рождения;
• половой член и мошонка резко гипоплазированы, отсутствуют их пубертатные изменения;
• вторичные половые признаки отсутствуют;
• типичные признаки проявляются в первые годы жизни – олигофрения, спастическая параплегия, пигментная ретинопатия.
Синдром Bardet – Biedl
Этиология: заболевание с аутосомно-рецессивным типом наследования. Выделяют три генетические формы: BBS1 картирован на 11q; BBS2 – 16q21; BBS3 – 3p.
Патогенез: не изучен.
Клинические проявления:
• двусторонний крипторхизм и микроорхидизм с рождения;
• половой член и мошонка резко гипоплазированы, отсутствуют их пубертатные изменения;
• вторичные половые признаки отсутствуют;
• типичны ожирение, олигофрения, полидактилия, пигментная ретинопатия.
Синдром Rod
Этиология: сцепленный с полом рецессивный или доминантный тип наследования.
Патогенез: не изучен.
Клинические проявления:
• часто встречается двусторонний крипторхизм и микроорхидизм с рождения;
• половой член и мошонка гипоплазированы, отсутствуют их пубертатные изменения;
• у большинства мальчиков клиника гипогонадизма появляется только в пубертатном возрасте, вторичные половые признаки отсутствуют или скудные;
• постоянный симптом – врожденный ихтиоз. Возможны умственная отсталость и эпилепсия.
Краниофарингиома
Эндокринные нарушения отмечаются примерно у 35 % пациентов.
Этиология: не изучена.
Патогенез:
• врожденная, как правило доброкачественная, опухоль головного мозга, развивающаяся из эмбриональных клеток гипофизарного хода (кармана Ратке);
• редкое заболевание, но самая частая супраселлярная опухоль детского возраста (5 – 10 % от опухолей головного мозга у детей);
• при отсутствии гормональной активности самой опухоли генез ее проявлений обусловлен механическим сдавливанием окружающих структур головного мозга, приводящим, в том числе, и к выпадению тропных функций гипофиза;
• изолированное выпадение гонадотропной функции встречается редко.
Клинические проявления:
• манифестация в большинстве случаев в детском и юношеском возрасте;
• как правило, сочетание симптомов внутричерепной гипертензии (головная боль, тошнота, рвота);
• хиазмальный синдром (битемпоральная гемианопсия, отек диска зрительного нерва, снижение остроты зрения);
• эндокринно-обменный синдром (гипопитуитаризм, задержка полового и физического развития);
• петрификаты над турецким седлом (65 – 75 %), выявляемые на рентгенограмме.
Гипоталамо-гипофизарная недостаточность (гипо-, пангипопитуитаризм)
Этиология: один из наследственных вариантов обусловлен мутациями гена PROP1, приводящими к нарушению дифференцировки клеток аденогипофиза, в том числе гонадотрофов. Непосредственные причины: травмы, хирургические повреждения, опухоли, инфекции.
Патогенез:
• деструктивные поражения гипоталамо-гипофизарной области, сопровождающиеся нарушением секреции гонадолиберина и (или) гонадотропных гормонов;
• выпадение других функций гипоталамо-гипофизарной области, в том числе гормональных и других тропных функций гипофиза.
Клинические проявления:
• в препубертатном возрасте – адипозогенитальная дистрофия с общим ожирением по «женскому» типу;
• евнухоидные пропорции тела, ложная гинекомастия;
• при врожденных формах недостаточности иногда бывает крипторхизм и микроорхидизм. В пубертатном возрасте нет тенденции к увеличению размера яичек;
• возможен микропенис и гипоплазия мошонки. Пубертатные изменения отсутствуют;
• скудное оволосение на лице, лобке и в подмышечных впадинах;
• возможен несахарный диабет;
• обязательные симптомы в детском возрасте – задержка роста с отставанием костного возраста.
Ятрогенные факторы, наркомания, алкоголизм
Этиология: лекарственные препараты, наркотики, этанол.
Патогенез:
• подавление продукции дофамина – основного ингибитора синтеза ПРЛ;
• развитие гиперпролактинемического гипогонадизма.
Клинические проявления:
• снижение либидо, импотенция;
• клинические проявления гипогонадизма;
• печеночно-почечная недостаточность.
Этиология: результат лечебно-диагностических воздействий.
Патогенез:
• нарушение участия почек в элиминации ПРЛ, находящегося в крови, – процесса, определяющегося в основном клубочковой фильтрацией гормона с его последующей канальцевой реабсорбцией и деградацией (Эммануэль Д. С. [и др.], 1987);
• снижение роли печени в инактивации и экскреции гормона;
• другие механизмы, связанные с воздействием различных этиологических факторов.
Проявления:
• снижение либидо, импотенция;
• гипогонадизм варьируется в большей степени и не имеет прямой связи с уровнем ПРЛ в крови.
НОРМОГОНАДОТРОПНЫЙ ГИПОГОНАДИЗМ
Микро– и макропролактинома
Гипофизарные опухоли из клеток аденогипофиза, продуцирующих пролактин.
Этиология: стресс + мутагенез.
Патогенез:
• изучается роль мутаций гена-супрессора Р-53 и G-белка в генезе пролактином;
• гиперсекреция пролактина, подавляющего продукцию ЛГ и ФСГ по короткой петле отрицательной обратной связи и тормозящего конверсиюТв5a-ДГТ;
• прямое влияние пролактина на центры головного мозга, снижающее половое влечение;
• механическое давление на мозговую ткань с выпадением гонадотропной функции гипофиза как в изолированном виде, такивсочетании с выпадением других тропных функций гипофиза.
Клинические проявления:
• в ряде случаев имеется задержка роста, микрогенитализм, отсутствие признаков полового развития в пубертатном периоде;
• у некоторых подростков микрогенитализм сочетается с ранним лобковым оволосением, высоким ростом, евнухоидностью и частым наличием гинекомастии;
• возможно сочетание гипогонадизма и ожирения с распределением жировой ткани по кушингоидному типу и розовыми стриями;
• большинство пациентов жалуются на слабость, избыточные прибавки массы тела, сонливость;
• исчезновение либидо;
• ослабление эрекции, импотенция;
• нарушение сперматогенеза;
• бесплодие;
• к моменту установления диагноза более 80 % пациентов имеют неврологические и зрительные нарушения как признак внутричерепной опухоли.
Первичный гипотиреоз
Этиология:
• наследственный (аутосомно-рецессивный) дефект в биосинтезе тиреоидных гормонов;
• гипо– и аплазия щитовидной железы;
• инфекционно-воспалительные и аутоиммунные поражения щитовидной железы;
• дефицит I2 (эндемический зоб), метастазы рака, хронические инфекции, лечение антитиреоидными препаратами.
Патогенез:
• длительный и выраженный дефицит гормонов щитовидной железы в результате их недостаточной секреции;
• повышенная секреция тиреотропин-рилизинг-гормона (ТРГ), как следствие нарушения механизма отрицательной обратной связи при дефиците тиреоидных гормонов;
• влияние повышенной концентрации ТРГ на секрецию пролактина гипофизом. ТРГ является мощным стимулятором выработки одновременно ТТГ и ПРЛ.
Клинические проявления:
• снижение либидо, импотенция;
• гипогонадизм.
Еще по теме 5.1. Гипогонадизм:
- 5.4. Лечение задержки полового развития и гипогонадизма
- 5.3. Лабораторные методы обследования при задержке полового развития и гипогонадизме
- Глава 5 ГИПОГОНАДИЗМ И ЗАДЕРЖКА ПОЛОВОГО РАЗВИТИЯ
- Глава 17 ЗАДЕРЖКИ ПОЛОВОГО РАЗВИТИЯ И МУЖСКОЙ ГИПОГОНАДИЗМ
- Нарушение функций половых желез
- БОЛЕЗНИ, АССОЦИИРОВАННЫЕ С БЕСПЛОДИЕМ.
- ПАТОГЕНЕЗ ГИПОГОНАДИЗМА У МУЖЧИН
- ГЕМОХРОМАТОЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ
- ETHINYLESTRADIOL (ЭТИНИЛЭСТРАДИОЛ)
- Синдром Клайнфельтера (47, XXY )
- Кахексия при СПИДе.
- Симптомы опухолей хиазмально-селлярной области.
- НЕИНФЕКЦИОННЫЕ ПРОСТАТИТЫ
- ПАТОГЕНЕЗ ГИПОГОНАДИЗМА У ЖЕНЩИН
- Атибактеріальна та протигрибкова терапія в педіатрії: Навчально-практичний посібник 11 видання / За ред. проф. В.В. Бережного. - Хмельницький,2016. - 416 с., 2016
- ТЕМА № 31 ГЕНЕРАЛИЗОВАННЫЕ ПОСЛЕРОДОВЫЕ ИНФЕКЦИОННЫЕ ЗАБОЛЕВАНИЯ ЛАКТАЦИОННЫЙ МАСТИТ СЕПТИЧЕСКИЙ ШОК В АКУШЕРСТВЕ
- ТЕМА № 30 ГНОЙНО-ВОСПАЛИТЕЛЬНЫЕ ПОСЛЕРОДОВЫЕ ЗАБОЛЕВАНИЯ
- ТЕМА № 29 НЕПРАВИЛЬНОЕ ПОЛОЖЕНИЕ ПЛОДА ОПЕРАЦИИ, ИСПРАВЛЯЮЩИЕ ПОЛОЖЕНИЯ ПЛОДА. АКУШЕРСКИЕ ПОВОРОТЫ ИЗВЛЕЧЕНИЕ ПЛОДА ЗА ТАЗОВЫЙ КОНЕЦ