оЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ
цНСООС ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ НАПЮГСЧР ГЮАНКЕБЮМХЪ, Б НЯґМНБЕ ЙНРНПШУ КЕФХР МЮЯКЕДЯРБЕММН НАСЯКНБКЕММЮЪ ДЕТЕЙРМНЯРЭ ЯРПСЙРСґПШ Х ТСМЙЖХНМХПНБЮМХЪ ХЛЛСММНИ ЯХЯРЕЛШ, ЙНРНПЮЪ ОПНЪБКЪЕРЯЪ Б МЮПСґЬЕМХХ ХЛЛСММНИ ГЮЫХРШ.
оЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ ≈ ЩРН НВЕМЭ ПЕДЙХЕ ЯНЯРНЪМХЪ (ОПХґЛЕПМН 1 АНКЭМНИ МЮ 1 000 000 ВЕКНБЕЙ). нМХ ЪБКЪЧРЯЪ ОНВРХ ХЯЙКЧВХРЕКЭґМН СДЕКНЛ ДЕРЯЙНЦН БНГПЮЯРЮ, ОНЯЙНКЭЙС ГМЮВХРЕКЭМЮЪ ВЮЯРЭ АНКЭМШУ Я РЪФЕКШЛХ ТНПЛЮЛХ ХЛЛСМНДЕТХЖХРНБ МЕ ДНФХБЮЕР ДН 20 КЕР, Ю ОПХ АНґКЕЕ КЕЦЙХУ ТНПЛЮУ ХЛЛСМНКНЦХВЕЯЙХЕ ДЕТЕЙРШ Я БНГПЮЯРНЛ Б НОПЕДЕКЕМґМНИ ЯРЕОЕМХ ЙНЛОЕМЯХПСЧРЯЪ.
йЮЙ ОПЮБХКН, Б НЯМНБЕ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ КЕФХР ЦЕМЕРХґВЕЯЙХ НАСЯКНБКЕММШИ АКНЙ ПЮГБХРХЪ ЙКЕРНЙ ХЛЛСММНИ ЯХЯРЕЛШ ХКХ БШОЮґДЕМХЕ БЮФМШУ ХЛЛСММШУ ОПНЖЕЯЯНБ БЯКЕДЯРБХЕ ДЕТЕЙРЮ НОПЕДЕКЕММШУ ЛНКЕЙСК, МЮОПХЛЕП ТЕПЛЕМРНБ ХКХ ЛЕЛАПЮММШУ ЯРПСЙРСП (ЯУЕЛЮ 4.3).
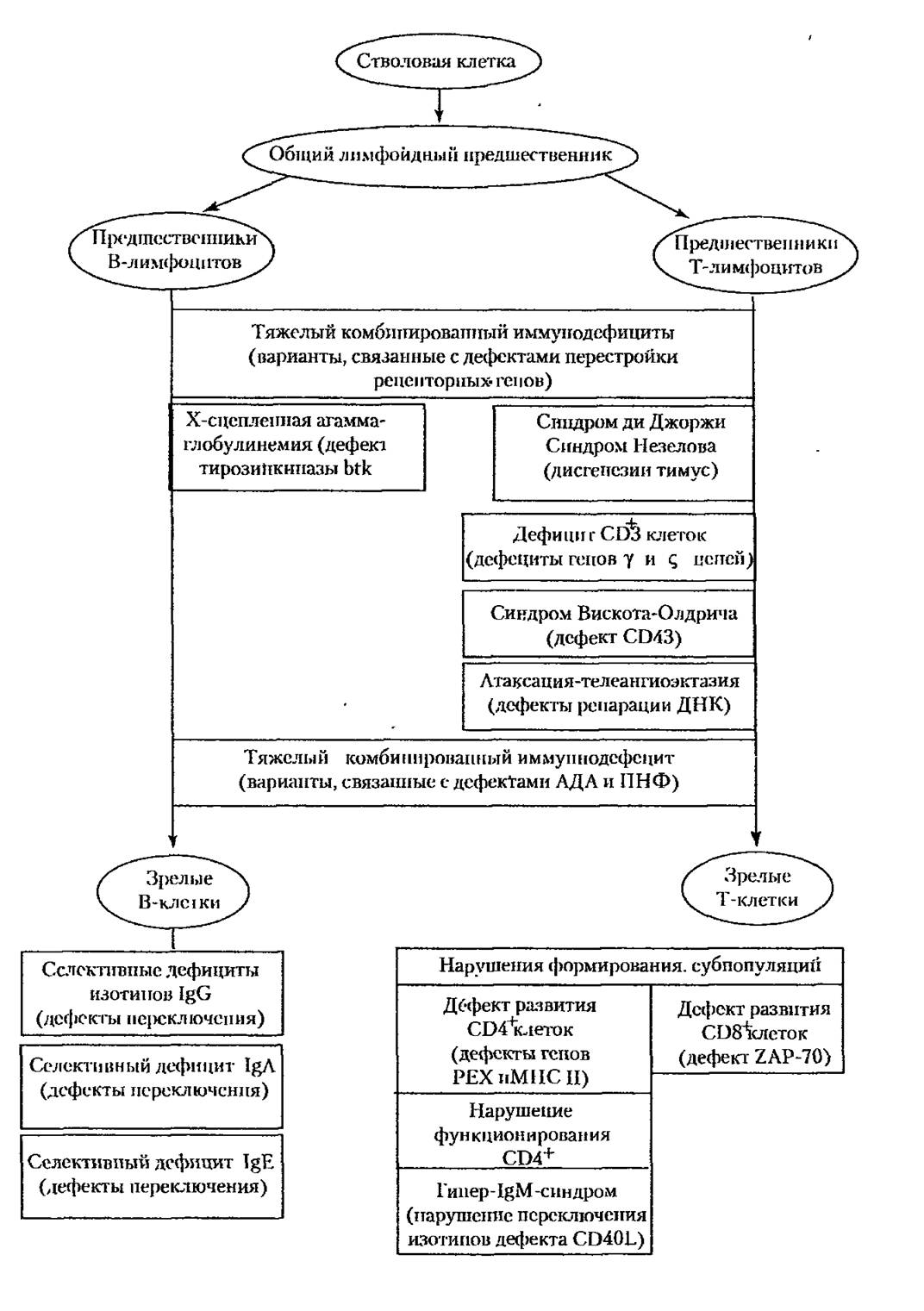
яУЕЛЮ 4.3. яБЪГЭ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ Я МЮПСЬЕМХЕЛ ПЮГБХРХЪ КХЛТНЖХРНБ МЮ НОПЕДЕКЕММШУ ЯРЮДХЪУ.
оЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ ЛНФМН ПЮГДЕКХРЭ ОН ОПЕНАКЮДЮЧЫЕЛС РХОС ОНПЮФЕМХИ ГБЕМЭЕБ ХЛЛСММНИ ЯХЯРЕЛШ МЮ 3 РХОЮ:
≈ ЙНЛАХМХПНБЮММШЕ ХЛЛСМНДЕТХЖХРШ;
≈ ХЛЛСМНДЕТХЖХРШ Я ОПЕХЛСЫЕЯРБЕММШЛ ОНПЮФЕМХЕЛ ЙКЕРНВМНЦН ХЛЛСМХРЕРЮ;
≈ ОПЕХЛСЫЕЯРБЕММН ЦСЛНПЮКЭМШЕ ХЛЛСМНДЕТХЖХРШ.
й ОЕПБШЛ НРМНЯЪР ГЮАНКЕБЮМХЪ, Б НЯМНБЕ ЙНРНПШУ КЕФЮР ЦЕМЕРХВЕЯґЙХЕ ДЕТЕЙРШ, ГЮРПЮЦХБЮЧЫХЕ ПЮГКХВМШЕ КХМХХ ДХТТЕПЕМЖХПНБЙХ КХЛґТНЖХРНБ, Ю РЮЙФЕ ПЮММХЕ ЩРЮОШ ХУ ПЮГБХРХЪ, НАЫХЕ ДКЪ р- Х б-КХМХИ. бН БРНПСЧ ЦПСООС БУНДЪР ХЛЛСМНДЕТХЖХРШ, ОПХ ЙНРНПШУ МЮПСЬЮЕРЯЪ ПЮГБХґРХЕ р-ЙКЕРНЙ Х ЯРПЮДЮЧР НОНЯПЕДСЕЛШЕ ХЛХ ПЕЮЙЖХХ ЙКЕРНВМНЦН ХЛЛСМХґРЕРЮ; Й ЩРНИ ФЕ ЦПСООЕ НРМНЯЪРЯЪ ДЕТЕЙРШ ТЮЦНЖХРХПСЧЫХУ ЙКЕРНЙ. б ЦПСОґОС ЦСЛНПЮКЭМШУ ХЛЛСМНДЕТХЖХРНБ БЙКЧВЮЧР ОЮРНКНЦХЧ, Б НЯМНБЕ ЙНРНПНИ КЕФХР МЮПСЬЕМХЕ ПЮГБХРХЪ б-ЙКЕРНЙ Х р-УЕКОЕПНБ ЦСЛНПЮКЭМНЦН НРБЕРЮ, Ю РЮЙФЕ ОЮРНКНЦХЧ ЙНЛОНМЕМРНБ ЙНЛОКЕЛЕМРЮ.
б ОНЯКЕДМХЕ ЦНДШ БШЪЯМЪЧРЯЪ ЛНКЕЙСКЪПМШЕ НЯМНБШ ОНПЮФЕМХЪ ОПХ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРЮУ. нДМНИ ХГ ОЕПБШУ АШКЮ ПЮЯЬХТПНБЮМЮ ОПХґПНДЮ ЙНЛАХМХПНБЮММШУ ХЛЛСМНДЕТХЖХРНБ, ЯБЪГЮММШУ Я МЕДНЯРЮРНВґМНЯРЭЧ ТЕПЛЕМРНБ ОСПХМНБНЦН ЛЕРЮАНКХГЛЮ. хГБЕЯРМШ БЮПХЮМРШ РЮЙХУ ДЕТЕЙРНБ, НАСЯКНБКЕММШЕ ЛСРЮЖХЪЛХ ЦЕМНБ, ЙНДХПСЧЫХУ ЮДЕМНГХМДЕГЮ- ЛХМЮГС Х ОСПХММСЙКЕНРХДТНЯТНПХКЮГС. нЯМНБНИ ДПСЦНИ ТНПЛШ РЪФЕКНґЦН ЙНЛАХМХПНБЮММНЦН ХЛЛСМНДЕТХЖХРЮ, ГЮРПЮЦХБЮЧЫЕЦН р- Х б-ПНЯРЙХ КХЛТНОНЩГЮ, ЯКСФХР ДЕТЕЙР ОПНЖЕЯЯЮ ОЕПЕЯРПНИЙХ ЦЕМНБ ЮМРХЦЕМПЮЯОНГґМЮЧЫХУ ПЕЖЕОРНПНБ, ЯБЪГЮММШИ Я НРЯСРЯРБХЕЛ ТЕПЛЕМРНБ ПЕЙНЛАХМЮГ, ЙНРНПШЕ ЙЮРЮКХГХПСЧР ЩРНР ОПНЖЕЯЯ.
нВЕМЭ ПЮГМННАПЮГЕМ ЯОЕЙРП ЦЕМЕРХВЕЯЙХ НАСЯКНБКЕММШУ МЮПСЬЕМХИ БШПЮАНРЙХ ЮМРХРЕК. хУ ОПХВХМНИ ЛНФЕР АШРЭ ЙЮЙ ОНПЮФЕМХЕ б-КХЛТНЖХґРНБ (ХУ ПЮГБХРХЪ ХКХ ЩЙЯОПЕЯЯХХ ЦЕМНБ ХЛЛСМНЦКНАСКХМНБ), РЮЙ Х ДЕТЕЙРґМНЯРЭ р-ЙКЕРНЙ (НЯКЮАКЕМХЕ УЕКОЕПМНИ ЮЙРХБМНЯРХ). оПХЛЕПНЛ ОЕПБНЦН ПНДЮ ЛНФЕР ЯКСФХРЭ ЮЦЮЛЛЮЦКНАСКХМЕЛХЪ аПСРНМЮ, ЯЖЕОКЕММЮЪ Я у-УПН- ЛНЯНЛНИ. еЕ НЯМНБНИ ЪБКЪЧРЯЪ ЛСРЮЖХХ ЦЕМЮ, ДЕРЕПЛХМХПСЧЫЕЦН ТЕПґЛЕМР РХПНГХМЙХМЮГС btk, ЙНРНПЮЪ ЯБЪГЮМЮ Я ЮМРХЦЕМПЮЯОНГМЮЧЫХЛ ПЕґЖЕОРНПНЛ б-КХЛТНЖХРНБ. нРЯСРЯРБХЕ ЩРНИ РХПНГХМЙХМЮГШ ДЕКЮЕР МЕБНГЛНФМШЛ ПЮГБХРХЕ б-КХЛТНЖХРНБ СФЕ МЮ ЯЮЛШУ ПЮММХУ ЯРЮДХЪУ.
б НЯМНБЕ ДПСЦНЦН ОЕПБХВМНЦН ХЛЛСМНДЕТХЖХРЮ ≈ ЦХОЕП-1Дл-ЯХМД- ПНЛЮ КЕФХР ДЕТЕЙР CD154 ≈ ЛНКЕЙСКШ, ОНЪБКЪЧЫЕИЯЪ МЮ ОНБЕПУМНЯРХ р-ЙКЕРНЙ ОПХ ХУ ЮЙРХБЮЖХХ; Б ПЕГСКЭРЮРЕ ЕЕ БГЮХЛНДЕИЯРБХЪ Я ЛНКЕЙСКНИ CD40 ОНБЕПУМНЯРХ б-КХЛТНЖХРНБ Б ЩРХ ЙКЕРЙХ ОЕПЕДЮЕРЯЪ ЯХЦМЮК, НАЕЯґОЕВХБЮЧЫХИ ХУ ДХТТЕПЕМЖХПНБЙС Б ЮМРХРЕКННАПЮГСЧЫХЕ ЙКЕРЙХ, Ю РЮЙґФЕ ОЕПЕЙКЧВЕМХЕ ХГНРХОНБ ЯЕЙПЕРХПСЕЛШУ ЮМРХРЕК. б НРЯСРЯРБХЕ ЩРНЦН ЯХЦМЮКЮ ОПНХЯУНДХР ЯХМРЕГ ХЛЛСМНЦКНАСКХМНБ РНКЭЙН НДМНЦН ХГНРХОЮ ≈ IgM, ВРН ЯНОПНБНФДЮЕРЯЪ НЯКЮАКЕМХЕЛ ЦСЛНПЮКЭМНЦН ХЛЛСММНЦН НРБЕРЮ. яСЫЕЯРБСЧР ТНПЛШ ЦСЛНПЮКЭМШУ ХЛЛСМНДЕТХЖХРНБ, ОПХ ЙНРНПШУ МЮПСґЬЕМН НАПЮГНБЮМХЕ ХЛЛСМНЦКНАСКХМНБ ЙЮЙНЦН-КХАН НДМНЦН ХГНРХОЮ. яПЕґДХ РЮЙХУ ЯЕКЕЙРХБМШУ ДЕТЕЙРНБ МЮХАНКЕЕ ВЮЯРШЛ ЪБКЪЕРЯЪ ДЕТХЖХР IgA.
оПХ МЕЛ ОПХЯСРЯРБСЧР б-КХЛТНЖХРШ, МЕЯСЫХЕ ЛЕЛАПЮММШИ IgA, НДМЮЙН МЕ НАПЮГСЧРЯЪ ОКЮГЛЮРХВЕЯЙХЕ ЙКЕРЙХ, ЯЕЙПЕРХПСЧЫХЕ IgA-ЮМРХРЕКЮ.
пЪД ЙНЛАХМХПНБЮММШУ ХЛЛСМНДЕТХЖХРНБ БНГМХЙЮЕР ОПХ КНЙЮКХГНґБЮММШУ ДЕТЕЙРЮУ ЦЕМНБ ЛЕЛАПЮММШУ ЛНКЕЙСК ЮДЦЕГХХ. яКЕДЯРБХЕЛ РЮЙХУ ЛСРЮЖХИ ЪБКЪЕРЯЪ МЮПСЬЕМХЕ ЛХЦПЮЖХХ ЙКЕРНЙ, Б ОЕПБСЧ НВЕПЕДЭ МЕИРґПНТХКНБ Х ЛНМНЖХРНБ/ЛЮЙПНТЮЦНБ, Ю РЮЙФЕ ХУ БГЮХЛНДЕИЯРБХИ Я ЙКЕРЙЮЛХ ДПСЦХУ РХОНБ. оПХЛЕПНЛ ЛНЦСР ЯКСФХРЭ ЯУНДМШЕ ОНПЮФЕМХЪ, ПЮГБХБЮЧґЫХЕЯЪ ЙЮЙ ПЕГСКЭРЮР МЮЯКЕДЯРБЕММШУ ДЕТЕЙРНБ ЩЙЯОПЕЯЯХХ (п2-ХМРЕЦПХ- МНБ Х СЦКЕБНДМШУ ДЕРЕПЛХМЮМР, ПЮЯОНГМЮБЮЕЛШУ ЯЕКЕЙРХМНЛ 1_. щРХ ОНПЮґФЕМХЪ ОПЕДЯРЮБКЪЧР ЯНАНИ ДБЮ БЮПХЮМРЮ LAD-ЯХМДПНЛЮ ≈- ДЕТХЖХР ЮДЦЕГХХ КЕИЙНЖХРНБ, ОПХГМЮЙНЛ ЙНРНПНЦН ЪБКЪЕРЯЪ НЯКЮАКЕМХЕ ТСМЙЖХХ МЕИРПНТХКНБ, Х ОНБШЬЕМХЕ ВСБЯРБХРЕКЭМНЯРХ Й ЦМНИМШЛ ХМТЕЙЖХЪЛ.
дЕТЕЙРШ ЙНЛОНМЕМРНБ ЙНЛОКЕЛЕМРЮ ОПЕДЯРЮБКЕМШ БЮПХЮМРЮЛХ Я ОНґПЮФЕМХЕЛ ОПЮЙРХВЕЯЙХ БЯЕУ НЯМНБМШУ ТЮЙРНПНБ ЙКЮЯЯХВЕЯЙНЦН Х ЮКЭРЕПґМЮРХБМНЦН ОСРЕИ ЮЙРХБЮЖХХ ЙНЛОКЕЛЕМРЮ. йЮЙ ОПЮБХКН, БШОЮДЕМХЕ ЕДХґМХВМШУ ЙНЛОНМЕМРНБ ЯХЯРЕЛШ ЙНЛОКЕЛЕМРЮ ОПНЪБКЪЕРЯЪ Б СЛЕПЕММНЛ ЯМХФЕМХХ СЯРНИВХБНЯРХ Й МЕЙНРНПШЛ-БНГАСДХРЕКЪЛ. кХЬЭ ДЕТХЖХР ХМЦХґАХРНПЮ я1 q ЯНОПНБНФДЮЕРЯЪ ПЮГБХРХЕЛ ЮМЦХНМЕБПНРХВЕЯЙНЦН НРЕЙЮ, НАСЯґКНБКЕММНЦН МЮЙНОКЕМХЕЛ БЮГНЮЙРХБМШУ ОЕОРХДНБ я5Ю Х ягЮ.
хЛЛСМНДЕТХЖХРШ, Б НЯМНБЕ ЙНРНПШУ КЕФХР ДЕТЕЙР ЦЕМНБ ЖХРНЙХМНБ, МЕЛМНЦНВХЯКЕММШ, ВРН ЯБЪГЮМН Я ╚ХГАШРНВМНЯРЭЧ╩ ЯХЯРЕЛШ ЖХРНЙХМНБ, ЙНРНПЮЪ НАСЯКНБКЕМЮ БГЮХЛНГЮЛЕМЪЕЛНЯРЭЧ ХУ ТСМЙЖХИ. кХЬЭ ЙНЦДЮ ЦЕґМЕРХВЕЯЙХИ ДЕТЕЙР ГЮРПЮЦХБЮЕР ТСМЙЖХЧ ЛМНЦХУ ЖХРНЙХМНБ, ЩРН ОПНЪБґКЪЕРЯЪ Б РЪФЕКШУ ПЮЯЯРПНИЯРБЮУ ХЛЛСМХРЕРЮ, ВРН ОПНХЯУНДХР, МЮОПХЛЕП, ОПХ ДЕТЕЙРЕ ЦЕМЮ С-ЖЕОХ, НАЫЕИ ДКЪ ПЕЖЕОРНПНБ ХМРЕПКЕИЙХМНБ 2, 4, 7, 13 Х 15.
б ПЕГСКЭРЮРЕ ДЕТЕЙРЮ, ГЮРПЮЦХБЮЧЫЕЦН ЦЕМ ЛЕЛАПЮММНЦН ЯХЮКНОПН- РЕХМЮ CD43, ПЮГБХБЮЕРЯЪ ЯХМДПНЛ бХЯЙНРРЮ≈нКДПХВЮ, Н ВЕЛ ЯБХДЕРЕКЭґЯРБСЕР РПНЛАНЖХРНОЕМХЪ Я ЦЕЛНППЮЦХВЕЯЙХЛ ЯХМДПНЛНЛ Б ЯНВЕРЮМХХ Я ЩЙГЕЛНИ Х ЙНЛАХМХПНБЮММШЛ ХЛЛСМНДЕТХЖХРНЛ. оПХ ЩРНЛ ГЮАНКЕБЮМХХ ЮМНЛЮКЭМН ТСМЙЖХНМХПСЕР ЖХРНЯЙЕКЕР, ВРН НРПЮФЮЕРЯЪ МЮ ОНДБХФМНЯРХ ЙКЕРНЙ Х ЛЕФЙКЕРНВМШУ БГЮХЛНДЕИЯРБХЪУ, БЮФМШУ ДКЪ НЯСЫЕЯРБКЕМХЪ ХЛЛСММШУ ОПНЖЕЯЯНБ.
оПХ ЮРЮЙЯХХ-РЕКЕЮМЦХЩЙРЮГХХ МЮАКЧДЮЕРЯЪ ОНПЮФЕМХЕ ПЮГКХВМШУ ТСМЙЖХИ, НАСЯКНБКЕММНЕ ЯКЮАНЯРЭЧ ЮООЮПЮРЮ ПЕОЮПЮЖХХ дмй Х МЕЯРЮґАХКЭМНЯРЭЧ УПНЛНЯНЛ, Ю РЮЙФЕ ДЕТЕЙРЮЛХ ЙКЕРНВМНЦН ЖХЙКЮ.
щРН ДЮЕР МЕНФХДЮММНЕ ЯНВЕРЮМХЕ ЯХЛОРНЛНБ: ЙНЛАХМХПНБЮММШИ ХЛЛСМНДЕТХЖХР (МЕДНПЮГБХРХЕ БХКНВЙНБНИ ФЕКЕГШ, ДЕТХЖХР р-ЙКЕРНЙ Х ХЛЛСМНЦКНАСКХґМНБ ╚ОНГДМХУ╩ ХГНРХОНБ ≈ lgG2, lgG4, IgE, IgA), МЕБПНКНЦХВЕЯЙХЕ НРЙКНґМЕМХЪ (ЮРЮЙЯХЪ), ОНПЮФЕМХЕ ЯНЯСДХЯРНИ ЯРЕМЙХ (РЕКЕЮМЦХЩЙРЮГХХ), МЮПСґЬЕМХЕ ОХЦЛЕМРЮЖХХ.оНЛХЛН ПЮЯЯЛНРПЕММШУ ╚РНВЕВМШУ╩ ОНПЮФЕМХИ ХЛЛСММНИ ЯХЯРЕЛШ ХГБЕЯРМШ ОЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ, ПЮГБХРХЕ ЙНРНПШУ НАСЯКНБКЕМН ЛМНФЕЯРБЕММШЛХ ДЕТЕЙРЮЛХ, ГЮРПЮЦХБЮЧЫХЛХ ТНПЛХПНБЮМХЕ Б ЩЛАПХґНЦЕМЕГЕ ПЮГКХВМШУ НПЦЮМНБ, БЙКЧВЮЪ НПЦЮМШ ХЛЛСММНИ ЯХЯРЕЛШ. рЮЙ. МЮЯґКЕДЯРБЕММШИ ОНПНЙ, ОПХБНДЪЫХИ Й МЮПСЬЕМХЧ ПЮГБХРХЪ С ЩЛАПХНМНБ 1п-
КНБЕЙЮ ОПНХГБНДМШУ 3 Х 4 ФЮАЕПМШУ ЫЕКЕИ, ЯКСФХР НЯМНБНИ ЯХМДПНЛЮ дХ дФНПДФХ Я ДЕТЕЙРНЛ ПЮГБХРХЪ БХКНВЙНБНИ ФЕКЕГШ (НМЮ МЕ ГЮЯЕКЪЕРЯЪ ОПЕДЬЕЯРБЕММХЙЮЛХ р-ЙКЕРНЙ, ПЮГБХРХЕ ЙНРНПШУ ОПЕПШБЮЕРЯЪ МЮ ЙНЯРМНґЛНГЦНБНИ ЯРЮДХХ) Х ЦХЯРНЦЕМЕРХВЕЯЙХ ПНДЯРБЕММШУ НПЦЮМНБ (ОЮПЮЫХРНґБХДМШУ ФЕКЕГ Х Р.Д).
нЯМНБМШЛ ЯХЛОРНЛНЙНЛОКЕЙЯНЛ, НРПЮФЮЧЫХЛ МЮПСЬЕМХЕ ХЛЛСМґМНИ ГЮЫХРШ ОПХ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРЮУ, ЪБКЪЕРЯЪ ХМТЕЙЖХНММШИ ЯХМДПНЛ, Р.Е. ОНМХФЕМХЕ ПЕГХЯРЕМРМНЯРХ Й ХМТЕЙЖХНММШЛ ЮЦЕМРЮЛ, Б РНЛ ВХЯКЕ ЯЮОПНТХРМШЛ (Pneumocystis carimi, Candida, ЖХРНЛЕЦЮКНБХПСЯ, МЕґЙНРНПШЕ ЩМРЕПНБХПСЯШ). уЮПЮЙРЕП МЮПСЬЕМХИ ХЛЛСММНИ ГЮЫХРШ НОПЕДЕґКЪЕРЯЪ КНЙЮКХГЮЖХЕИ ОНПЮФЕМХЪ Б ХЛЛСММНИ ЯХЯРЕЛЕ. рЮЙ, ОПХ АКНЙЮДЕ ОПНЖЕЯЯЮ ОЕПЕЯРПНИЙХ ПЕЖЕОРНПМШУ ЦЕМНБ НРЯСРЯРБСЧР ЙЮЙ р-, РЮЙ Х б-ЙКЕРґЙХ Х МЕ ПЮГБХБЮЧРЯЪ МХ ЙКЕРНВМШЕ, МХ ЦСЛНПЮКЭМШЕ ТНПЛШ ХЛЛСММНЦН НРБЕРЮ. оПХ ЯЕКЕЙРХБМШУ ДЕТЕЙРЮУ НОПЕДЕКЕММШУ ЙКЮЯЯНБ КХЛТНЖХРНБ, Ю РЮЙФЕ ХУ ЯСАОНОСКЪЖХИ БШОЮДЮЧР ХЛЕММН РЕ ХЛЛСМНКНЦХВЕЯЙХЕ ТСМЙЖХХ, ГЮ ЙНРНПШЕ НРБЕРЯРБЕММШ ОНПЮФЮЕЛШЕ РХОШ ЙКЕРНЙ. оПХ АКНЙЮДЕ ПЮГБХґРХЪ б-ЙКЕРНЙ ПЮГБХБЮЕРЯЪ ЮЦЮЛЛЮЦКНАСКХМЕЛХЪ Я МЮПСЬЕМХЕЛ ЦСЛНПЮКЭґМНИ ГЮЫХРШ НР БМЕЙКЕРНВМШУ АЮЙРЕПХИ Х ХУ РНЙЯХМНБ, Ю ОПХ ДЕТХЖХРЮУ р-КХЛТНЖХРНБ ЯРПЮДЮЕР ЙКЕРНВМЮЪ ГЮЫХРЮ НР БХПСЯНБ Х ЛХЙНАЮЙРЕПХИ. оПХ МЕЙНРНПШУ ТНПЛЮУ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ (ЮРЮЙЯХЪ-РЕКЕЮМЦХЩЙЯРЮ- ГХЪ, ЯХМДПНЛ бХЯЙНРРЮ-нКДПХВЮ Х Р.Д.) ГМЮВХРЕКЭМН ОНБШЬЮЕРЯЪ ПХЯЙ ПЮГґБХРХЪ ГКНЙЮВЕЯРБЕММШУ НОСУНКЕИ (ДН 10≈15%). мЕПЕДЙН МЮПСЬЕМХЪ ХЛґЛСМНКНЦХВЕЯЙХУ ТСМЙЖХИ ПЕЦХЯРПХПСЧРЯЪ ОПХ МНПЛЮКЭМНИ ВХЯКЕММНЯРХ ЯННРБЕРЯРБСЧЫХУ ЙКЕРНЙ.
йКХМХЙН-ХЛЛСМНКНЦХВЕЯЙНЕ НАЯКЕДНБЮМХЕ ДЮЕР ВЕРЙХЕ ПЕГСКЭРЮРШ КХЬЭ ОПХ РЕУ ТНПЛЮУ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ, ОПХ ЙНРНПШУ РНВМН КНЙЮКХГНБЮМ ДЕТЕЙР. рЮЙ, ОПХ РЪФЕКНЛ ЙНЛАХМХПНБЮММНЛ ХЛЛСМНДЕТХґЖХРЕ НРЯСРЯРБСЧР ЙЮЙ р-, РЮЙ Х б-ЙКЕРЙХ, ОПХ ЯХМДПНЛЕ дХ дФНПДФХ ПЕГЙН ЯМХФЕМН ЯНДЕПФЮМХЕ р-КХЛТНЖХРНБ, Ю ОПХ ЮЦЮЛЛЮЦКНАСКХМЕЛХЪУ ≈ б-КХЛТНЖХРНБ. оН ХГЛЕМЕМХЧ ЙНМЖЕМРПЮЖХХ ХЛЛСМНЦКНАСКХМНБ Б ЯШБНґПНРЙЕ ХКХ ЙНЛОНМЕМРНБ ЙНЛОКЕЛЕМРЮ ПЮГКХВМШУ ХГНРХОНБ ЛНФЕР АШРЭ СЯґРЮМНБКЕМЮ КНЙЮКХГЮЖХЪ ДЕТЕЙРЮ Б ЯХЯРЕЛЕ ЦСЛНПЮКЭМНЦН ХЛЛСМХРЕРЮ. бЯЕ АНКЭЬСЧ ДХЮЦМНЯРХВЕЯЙСЧ ГМЮВХЛНЯРЭ ОПХНАПЕРЮЕР НОПЕДЕКЕМХЕ ЙНМЙґПЕРМШУ ЛЕЛАПЮММШУ ЛЮПЙЕПНБ ЙКЕРНЙ ХЛЛСММНИ ЯХЯРЕЛШ (ЛНКЕЙСК ЮДЦЕґГХХ, CD154, CD43 Х Р.Д.), Ю РЮЙФЕ ЛЕРНДШ, ОНГБНКЪЧЫХЕ БШЪБХРЭ ЛСРЮЖХХ ЙНМЙПЕРМШУ ЦЕМНБ.
4.3.2.
еЫЕ ОН РЕЛЕ оЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ:
- цКЮБЮ 18. оЕПБХВМШЕ ХЛЛСМНДЕТХЖХРШ
- 4.1.3. оНДУНДШ Й РЕПЮОХХ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ.
- 4.1.4. нАЫХЕ ОПХМЖХОШ КЕВЕМХЪ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ.
- 4.1.1. пЮАНВЮЪ ЙКЮЯЯХТХЙЮЖХЪ ОЕПБХВМШУ ХЛЛСМНДЕТХЖХРНБ.
- кЕВЕМХЕ АНКЭМШУ ОЕПБХВМШЛ ЯЕПНМЕЦЮРХБМШЛ, ОЕПБХВМШЛ ЯЕПНОНГХґРХБМШЛ Х БРНПХВМШЛ ЯБЕФХЛ ЯХТХКХЯНЛ.
- бРНПХВМШЕ ХЛЛСМНДЕТХЖХРШ
- оПХ ОНДНГПЕМХХ МЮ ОЕПБХВМШИ АХКХЮПМШИ ЖХППНГ ОЕВЕМХ ХКХ ОЕПБХВМШИ ЯЙКЕПНГХПСЧЫХИ УНКЮМЦХР
- 4.2.2. йКЮЯЯХТХЙЮЖХЪ БРНПХВМШУ ХЛЛСМНДЕТХЖХРНБ.
- брнпхвмши (опхнапереммши) хллсмндетхжхр
- яХМДПНЛ ОПХНАПЕРЕММНЦН ХЛЛСМНДЕТХЖХРЮ (яохд) ВЕКНБЕЙЮ
- бпнфдеммши (оепбхвмши) хллсмндетхжхр
- 4.2.1. щРХНКНЦХЪ БРНПХВМШУ ХЛЛСМНДЕТХЖХРНБ.
- бРНПХВМШЕ (ОПХНАПЕРЕММШЕ) ХЛЛСМНДЕТХЖХРШ МЮАКЧДЮЧРЯЪ:
- 4.1 гЮАНКЕБЮМХЪ √ ЛЮПЙЕПШ ХЛЛСМНДЕТХЖХРЮ С НАЯКЕДНБЮММШУ АНКЭМШУ
- цкюбю 27.3 оЕПБХВМЮЪ ОЕПХРНМЕЮКЭМЮЪ ЙЮПЖХМНЛЮ (оЕПБХВМШИ ОЕПХРНМЕЮКЭМШИ ПЮЙ) (я48.1, я48.2, я48.8)
- оЕПБХВМЮЪ ОЕПХРНМЕЮКЭМЮЪ ЙЮПЖХМНЛЮ (оЕПБХВМШИ ОЕПХРНМЕЮКЭМШИ ПЮЙ) (я48.1, я48.2, я48.8)
- ю. оЕПБХВМШИ ЙНЛОКЕЙЯ
- оЕПБХВМШЕ ДХЯРПНТХХ
- оЕПБХВМШИ ЯХТХКХЯ