ПАТОГЕНЕЗ НАИБОЛЕЕ ИЗУЧЕННЫХ ВРОЖДЕННЫХ ИММУНОДЕФИЦИТОВ НА УРОВНЕ ИММУНОКОМПЕТЕНТНЫХ КЛЕТОК
Расстройства дифференциации общего предшественника лимфоидных клеток приводят к блокаде образования Т- и В-лимфоцитов, что обуславливает тяжелую недостаточность системы иммунитета реакции в ответ на ее стимуляцию антигенами (табл.
30.1.). При этом из организменной реакции
Врожденные иммунодефициты
 Иммунодефициты в результате недостаточности и расстройств реакции на антигенную стимуляцию на уровне им му некомпетентных клеток
Иммунодефициты в результате недостаточности и расстройств реакции на антигенную стимуляцию на уровне им му некомпетентных клеток
Некоторые из нозологических форм

 Ретикулярный дисгенез, тяжелый комбинированный дефицит с аплазией вилочковой железы, частый вариабельный иммунодефицит при задержке роста тела, агамма-глобулинемия, связанная с Х-хромосомой
Ретикулярный дисгенез, тяжелый комбинированный дефицит с аплазией вилочковой железы, частый вариабельный иммунодефицит при задержке роста тела, агамма-глобулинемия, связанная с Х-хромосомой
Болезнь Брутона и другие виды врожденной гипогамма-глобули- немии, иногда сочетающиеся с тимомой, частый вариабельный иммунодефицит
Болезнь Незелофа, синдром ДиДжорджи, дефицит нуклеозид- фосфорилазы
Синдром Вискотта-Олдрича, иммунодефицит при атаксии- тел еанги эктазии
 Нарушения на уровне молекулярных иммунных систем и неспецифических клеток системы иммунитета
Нарушения на уровне молекулярных иммунных систем и неспецифических клеток системы иммунитета
Некоторые из нозологических форм и патологических состояний
 Нейтропения, синдром ленивых лейкоцитов
Нейтропения, синдром ленивых лейкоцитов
Сниженная бактерицидная активность фагоцитов при хронических гранулематозных заболеваниях, дефиците миелопероксидазы, синдроме Чедиака-Хигаси (нейтрофилы и мононуклеары с дефектными лизосомами, недостаточной способностью к хемотаксису и высвобождению бактерицидных веществ), дефиците активности глюкозо-6-фосфат-дегидрогеназы
Ангионевротический отек (аномалии на уровне ингибитора С1-эстеразы), системная красная волчанка
системы иммунитета выпадает работа клеточных и молекулярных эффекторов систем ее регуляции (Т-индукторы, Т-супрессоры и Т-хелперы, а также их цитокины) и клеток, через активацию которых происходит уничтожение и элиминация чужеродных иммуногенов и антигенов (В-клетки, плазматические клетки, Т-лимфоциты натуральные киллеры и цитотокси- ческие Т-лимфоциты).
Кроме того, при тяжелом иммунодефиците вследствие недостаточности стволовых клеток неэффективен эндоцитоз моно- и полиморфонуклеарами, что отчасти связано с критическим дефицитом иммуноглобулинов в качестве опсонинов.У некоторых больных недостаточность дифференциации клеток предшественниц лимфоцитов является результатом недостаточной активности аденозиндезаминазы. У 50% таких пациентов к положительному результату приводит переливание нормальных эритроцитов, содержащих данный фермент. При длительном течении заболевания, когда иммунодефицит сопровождается атрофией эпителия вилочковой железы, требуется дополнительное лечение экстрактами нормальной вилочковой железы (тимозин и
др.). Тяжелый комбинированный иммунодефицит, вызванный полным отсутствием предшественников не только иммунокомпетентных, но и всех миелоидных клеток, получил название ретикулярного дисгенеза. Заболевание приводит к смерти вскоре после рождения.
У детей с недостаточностью стволовых клеток нормальная реактивность иммунной системы может быть восстановлена трансплантацией гистосов- местимого с организмом донора костного мозга от его брата или сестры.
В-лимфоцит - это иммунокомпетентная клетка, образуемая сумкой Фабрициуса, органом, локализация которого у млекопитающих до сих пор не выяснена. В-лимфоциты составляют 30 % всех циркулирующих с кровью лимфоцитов и сконцентрированы в фолликулярной зоне лимфатических узлов. В-лимфоциты образуют иммуноглобулины, таким образом участвуя:
♦ в системной защитной реакции, направленной против патогенных микроорганизмов;
♦ в иммунном надзоре за клетками своего организма, то есть в цитолизе потенциально злокачественных клеток;
♦ в антитело-зависимой клеточной реакции цитотоксичности и реакциях повышенной чувствительности.
Как антитело-продуцирующие клетки В-лимфоциты образуют комплексы антиген-антитело. При образовании комплексов на поверхности объектов фагоцитоза иммуноглобулины начинают функционировать в качестве опсонинов, что повышает эффективность защитной реакции фагоцитоза.
Образование на поверхности объектов фагоцитоза комплексов антиген-ан- титело активирует систему комплемента по классическому пути. Активация системы комплемента служит инициирующим моментом воспаления. Поэтому недостаточность образования В-клетками иммуноглобулинов приводит к недостаточности воспаления как защитной реакции, направленной на элиминацию носителей чужеродных антигенов. В-лимфоциты, секрети- рующие широкий спектр цитокинов, - необходимый эффектор системы регуляции системной иммунной реакции. В клетках-предшественниках В-лимфоцитов есть цитоплазматические иммуноглобулины. При этом поверхность клеток-предшественников иммуноглобулинов не содержит. Маркерами зрелости В-лимфоцитов являются антигены их наружной клеточной поверхности и цитоплазмы. Зрелые В-лимфоциты несут на наружной поверхности иммуноглобулины и рецепторы фракций системы комплемента. Антигены- маркеры зрелости (нормальной окончательной дифференциации) В-лимфоцитов - это СЭ9, СОЮ, СО 19, С020, С024, Рс-рецептор, В1, ВА-1, В4 и 1а.Связанная с Х-хромосомой агамма-глобулинемия (болезнь Брутона) - первичный иммунодефицит, в основе которого лежит задержка нормальной дифференциации В-лимфоцитов. Болезнь Брутона встречается только у мужчин. Вследствие такой задержки у больного младенца нет своих зрелых В-лимфоцитов и эндогенных гамма-глобулинов плазмы крови. Заболевание начинает проявлять себя с шестимесячного возраста, в котором из плазмы крови ребенка элиминируются иммуноглобулины матери. В результате иммунодефицита, несмотря на нормальные свойства Т-лимфоцитов, у больного возникают очаги гнойной инфекции разной локализации. Имму
нодефицит усиливается вследствие синдрома нарушенного кишечного всасывания, обычно связанного с лямблиозом. В частности, синдром составляют полиартрит, системный коллагеноз, вызывающий распространенный васкулит, а также дерматомиозит, который у части больных приводит к летальному исходу.
Транзиторная гипогамма-глобулинемия - иммунодефицит, схожий в своих проявлениях с болезнью Брутона и обусловленный замедленным становлением иммунной системы у новорожденных.
В результате после элиминации иммуноглобулинов матери из плазмы ребенка в ней некоторое время нет зрелых В-лимфоцитов и достаточной концентрации гамма-глобулинов.Связанная с полом гипогаммаглобулинемия при нормальном или повышенном содержании иммуноглобулинов М в плазме крови. Этот первичный иммунодефицит характеризуют дефицит иммуноглобулинов в и А при нормальной или повышенной концентрации в плазме крови иммуноглобулинов М. У больных в плазме крови выявляют В-клетки, способные образовывать и нести на своей поверхности иммуноглобулины М. Грубых дисфункций Т-лимфоцитов нет. В основе синдрома лежит задержка трансформации продуцирующих иммуноглобулины М В-клеток в более зрелую форму В-лимфоцитов, образующую иммуноглобулины в. Большинство больных - женщины. Синдром часто возникает одновременно с нейтропе- нией, тромбоцитопенией, гемолитической анемией и В-клеточной лимфо- мой. Приобретенный иммунодефицит с аналогичным патогенезом вызывают краснуха, недостаток транскобаламина II. Врожденный и наследуемый по аутосомно-рецессивному типу недостаток в организме транскобаламина II (транспортный белок, вовлеченный в метаболизм витамина В|2), может приводить к полному отсутствию в плазме крови гамма-глобулинов, агам- маглобулинемии. Врожденный недостаток витамина В|2 ведет к мегалобла- стической анемии, гранулоцитопении и тромбоцитопении, снижающим общую резистентность организма, что вкупе с иммунодефицитом делает прогноз особенно неблагоприятным.
Тяжелый комбинированный иммунодефицит - весьма вариабельное в своих клинических проявлениях врожденное иммунопатологическое состояние, которое наследуется по аутосомно-рецессивному типу. Кроме того, наследование тяжелого комбинированного иммунодефицита может быть связано с Х-хромосомой. В Соединенных Штатах этот иммунодефицит чаще выявляют у черных младенцев в возрасте до пяти месяцев. В основе тяжелого комбинированного иммунодефицита лежат дисфункции как В-, так и Т-лимфоцитов. Исследование иммунологического статуса больных выявляет:
♦ сниженную концентрацию иммуноглобулинов в плазме крови или их полное в ней отсутствие;
♦ нарушение клеточного звена иммунитета, которое проявляет себя полным угнетением реакции интенсификации митоза Т-клеток в ответ на действие специфических для них митогенов (фитогемагглю- тинина и др.);
♦ снижение содержания в крови как В-, так и Т-лимфоцитов;
♦ угнетения образования интерлейкина-2 клетками иммунной системы.
Тяжелый комбинированный иммунодефицит вызывают:
♦ наследственная недостаточность энзима аденозиндеаминазы;
♦ врожденный дефицит энзима пуриннуклеозидфосфорилазы;
♦ генетически детерминированный дефект белков, которые, связываясь с дезоксирибонуклеиновой кислотой, определяют экспрессию генов из главного комплекса тканевой совместимости (система антигенов лейкоцитов человека).
Кроме того, тяжелый комбинированный иммунодефицит считают вариантом болезни Незелофа.
Клинические проявления тяжелого комбинированного иммунодефицита складываются из сыпи, похожей на сыпь у больных корью, а также частых легочных инфекций (Candida, Pneumocystis carinii, вирус Эпштейн-Барра, возбудитель ветряной оспы и др.). Нередкой является гиперпигментация кожи.
Частый вариабельный иммунодефицит (англ. - common variable immunodeficiency) - это наследуемое по аутосомно-рецессивному типу нарушение реакции системы иммунитета организма в ответ на антигенную стимуляцию, которое характеризует снижение концентрации в плазме крови иммуноглобулинов большинства изотипов. При вариабельном иммунодефиците клетки-предшественники В-лимфоцитов циркулируют с кровью, но не дифференцируются до зрелых форм. Кроме того, у больных выявляют дефекты развития Т-клеток, которые не служат первопричиной расстройств клеточного звена иммунитета. Частым этот вид врожденного комбинированного иммунодефицита называют потому, что его частота среди всех видов первичного иммунодефицита высока и составляет 20-90 случаев на миллион населения.
Заболевания и синдромы, связанные с частым вариабельным иммунодефицитом, начинают выявлять с двенадцатилетнего возраста. Их спектр широк. В этой связи это иммунопатологическое состояние получило название вариабельного. Чаще всего больные с вариабельным иммунодефицитом страдают от различных бактериальных инфекций, вызывающих отиты, синуситы, пневмонии. Нередко их беспокоят поносы неизвестного происхождения, которые выступают основным симптомом синдрома нарушенного кишечного всасывания у таких пациентов.
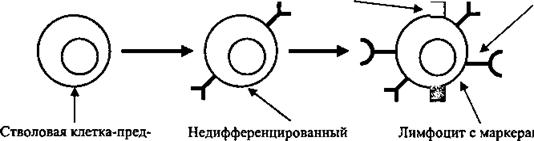
Синдром нарушенного кишечного всасывания часто сопровождается энтеритом, ахлоргидрией и ведет к пернициозной анемии. Синдром нарушенного кишечного всасывания у больных вызывается и холестазом той или иной степени, в частности, связанным с лямблиозом.Основное звено патогенеза вариабельного иммунодефицита - это блокирование дифференцировки клеток-предшественников В-лимфоцитов до зрелых форм (рис. 30.1). Предположительно одним из этиологических факторов вариабельного иммунодефицита считают вирус Эпштейна-Барра. Его патогенные эффекты могут резко угнетать образование иммуноглобулинов зрелыми В-лимфоцитами, нарушать гпикозилирование антител или воздействовать на Т-лимфоциты таким образом, что они начинают подавлять синтез иммуноглобулинов.
| Рс-рецептор СЗ-рецептор
шественница иммуно- лимфоцит с поверхностными клеточной дифференциации компетентных клеток иммуноглобулинами на клеточной поверхности Рис. 30.1. Принципиальная схема дифференциации В-лимфоцитов |
Исследование плазмы крови больных с вариабельным частым иммунодефицитом позволяет выявить значительную гипогаммаглобулинемию. Из результатов специальных исследований больных с данным иммунодефицитом можно выделить снижение активности 5‘-нуклеотидазы в лимфоцитах пациентов с данным видом иммунодефицита. У таких больных риск развития рака желудка в 50 раз выше, чем у представителей основной популяции. Активность этого фермента снижена в лимфоцитах у 70 % больных с частым вариабельным иммунодефицитом. Падение секреции гастрина под влиянием бомбезина после инъекции пептида является маркером частого комбинированного иммунодефицита у больных с высоким риском рака желудка.
Врожденные иммунодефициты, связанные с расстройствами системной иммунной реакции исключительно на уровне Т-лимфоцитов, у больных с недостаточной реакцией системы иммунитета в ответ на антигенную стимуляцию встречаются весьма редко. Врожденные дефекты свойств Т-лим- фоцитов почти всегда приводят к недостаточному образованию антител В- лимфоцитами. Дело в том, что большинство белковых антигенов и гапте- нов обладают свойством стимулировать систему иммунитета для образования антител лишь при условии ненарушенного функционального состояния как Т-лимфоцитов, так и В-клеток. Полное отсутствие в организме нормальных Т-лимфоцитов лишает его систему иммунитета способности реагировать образованием антител в ответ на стимуляцию Т-лимфоцит-за- висимыми антигенами. В этой связи можно считать, что вне зависимости от первопричины недостаточной реакции системы иммунитета в виде избирательного дефекта В- или Т-лимфоцитов, иммунодефицит на уровне имму- нокомпетентных клеток всегда отчасти является комбинированным, то есть его патогенез состоит из дисфункций иммунной системы как на уровне Т- клеток, так и В-лимфоцитов.
Т-лимфоциты участвуют в защитных реакциях, направленных на элиминацию из организма возбудителей внутриклеточных инфекций. Каждая Т-клетка имеет специфический рецептор, благодаря которому и распознает антиген во взаимодействии с мононуклеаром. Т-лимфоцит распознает антигены возбудителей внутриклеточных инфекций в комплексе с молекулами из главного комплекса тканевой совместимости на поверхности антиген-презентирующей клетки. В результате образования комплекса, состоящего из Т-клеточного рецептора и антигена, происходит индукция клональной экспансии лимфоцитов с образованием эффекторных клеток и клеток памяти, что обеспечивает специфическую реакцию приобретенного иммунитета.
Т-хелперы распознают антигены возбудителей внутриклеточных инфекций, экспонированные на поверхности мононуклеарных фагоцитов в комплексе с молекулами второго класса из главного комплекса тканевой совместимости (ГКТС). После распознавания Т-хелперы секретируют цитокины, и в частности гамма-интерферон. Через секрецию цитокинов хелперы действуют как клеточные эффекторы регуляции системной иммунной реакции. В результате их действия в данном качестве активируются цитотоксические лимфоциты и натуральные киллеры, которые уничтожают инфицированные клетки. Цитотоксические Т-лимфоциты распознают антигены возбудителей внутриклеточных инфекций, которые экспонированы на поверхности атакуемых клеток в комплексе с гликопротеинами из первого класса молекул ГКТС.
Поэтому недостаточность системной иммунной реакции, связанная преимущественно с дисфункциями системы иммунитета на уровне Т-лимфо- цитов, приводит к частым болезням, в основе которых лежат вирусные, бактериальные и паразитарные внутриклеточные инфекции.
Диагноз врожденной аплазии вилочковой железы (синдромы ДиДжорд- жи, Незелофа) становится ясным вскоре после рождения, так как синдром как патологическое состояние всего организма включает врожденные пороки сердца и первичный гипопаратиреоз. В результате гипопаратиреоза у таких больных уже через 24 часа после рождения развивается гипокальци- емия как причина тетании, то есть перемежающихся судорог. Синдром ДиД- жорджи представляет собой результат нарушения эмбрионального развития на двенадцатой неделе беременности, когда не происходит нормального формирования вилочковой и паращитовидных желез из третьего и четвертого парных выпячиваний эмбриональной глоточной эктодермы. У многих больных нет полной аплазии железы; она лишь частична при нередкой аномальной локализации. Из-за недоразвития вилочковой железы стволовые клетки не могут дифференцироваться в Т-лимфоциты, и «тимус-зависимые» элементы лимфоидной ткани организма характеризует пониженное содержание лимфоцитов. В результате у больных выявляют патологически низкое содержание Т-лимфоцитов в циркулирующей крови. Если больные выживают, несмотря на сердечную недостаточность вследствие врожденного порока сердца, то причиной летального исхода служат осложнения соответствующих инфекционных заболеваний. Дело в том, что у таких больных практически нет нормальной реакции клеточного иммунитета, хотя их иммунная система способна уничтожать возбудителей бактериальных инфекций. Система иммунитета у больных с синдромами ДиДжорджи и Незелофа обладает способностью продуцировать антитела в ответ на стимуляцию антигенами, но эта реакция ослаблена, так как системную иммунную реакцию не обеспечивает должным образом взаимодействие Т- и В-лимфоци-
тов. У больных с аплазией вилочковой железы «... иммунореактивность восстанавливается при трансплантации тимуса новорожденных, но, если трансплантат подобран неудачно, он отторгается “неблагодарными” клетками хозяина, созреванию которых сам же и способствовал» (Ройт).
Синдром Вискота-Олдрича относится к первичным иммунодефицитам вследствие выпадения из системной иммунной реакции нормальной работы как Т-, так и В-лимфоцитов. В частности, синдром характеризует тром- боцитопения, которую у части больных выявляют сразу после рождения. Наследование синдрома связано с полом. Его возникновение определяет патогенный рецессивный аллель гена болезни. Первопричина болезни состоит в патологическом изменении функционального состояния мононук- леарных фагоцитов, которое определяет невозможность индукции системной иммунной реакции через взаимодействие антигенпрезентирующей клетки и Т-лимфоцита. Дело в том, что мононуклеарные фагоциты у больных с синдромом не обладают способностью переработки для представления иммуноцитам полисахаридных антигенов. В результате система иммунитета не обладает способностью вырабатывать антитела, комплементарные по отношению к полисахаридным антигенам некоторых патогенных микроорганизмов {Hemophilus influenzae, пневмококки и др.). В данном случае первопричина иммунодефицита на уровне мононуклеарных фагоцитов определяет вторичную специфическую недостаточность антителообразования В-клетками. Если сначала по ходу онтогенеза исследование функций имму- нокомпетентных клеток in vitro не выявляет аномалий, то затем они возникают. Больные с синдромом часто страдают от повышенной кровоточивости, связанной с тромбоцитопенией, подвержены малигнизации клеток лимфоретикулоэндотелиальной системы и частым инфекционным заболеваниям, которые вызывают соответствующие возбудители.
Иммунодефицит у больных с атаксией-телеангиэктазией. Атаксия - неспособность координации мышц при произвольных движениях. Телеанги- эктазия - локальное расширение капилляров и мелких концевых артерий. Атаксия-телеангиэктазия представляет собой моногенную болезнь, наследуемую по аутосомно-рецессивному типу. Кроме атаксии и телеангиэкта- зии болезнь характеризуют частые или хронические инфекции. Примерно у 80% больных в сыворотке крови и секретах почти нет иммуноглобулинов А. У большинства пациентов патологически снижена не только концентрация иммуноглобулинов А в секретах и сыворотке, но и содержание в них иммуноглобулинов G. Болезнь в морфопатогенетическом отношении характеризуют аутоантитела к иммуноглобулинам А, гипоплазия вилочковой железы, лимфопения, а также сниженная реакция Т-лимфоцитов в ответ на эффект митогенов. Предположительно основным звеном патогенеза иммунодефицита у больных с атаксией-телеангиэктазией можно считать недостаточные влияния со стороны Т-лимфоцитов на развитие В-клеток или дефицит их регуляторных влияний в качестве супрессоров.
Иммунодефищты как следствия врожденного недостатка активности аденозиндезаминазы и нуклеозидфосфорилазы. Если врожденный дефицит активности аденозиндеаминазы служит причиной иммунодефицита вследствие патологического изменения функционального состояния как Т-, так и В-лимфоцитов, то наследственный недостаток активности нуклеозид- фосфорилазы обуславливает иммунодефицит преимущественно на уровне Т-клеток.
Первичные одновременные нарушения дифференциации как В-, так и Т-лимфоцитов, приводящие к врожденному комбинированному иммунодефициту, связаны с наследуемым по аутосомно-рецессивному типу недостатком фермента аденозиндеаминазы. Ген энзима локализован на длинном плече 22 аутосомы. Сам фермент представляет собой молекулу с массой в 38 килодальтон. Дефицит энзима ведет к накоплению в клетках аденози- на, деоксиаденозина, аденозинтрифосфата, Б-аденозилгомоцистеина и деоксиаденозинтрифосфорной кислоты. Деоксиаденозинтрифосфат - это мощный естественный ингибитор фермента дифосфатредуктазы, который участвует в синтезе пуринов. Рост в клетках концентрации аденозина угнетает внутриклеточное метилирование дезоксирибонуклеиновой кислоты (ДНК), что служит причиной цитолиза. Кроме того, врожденный недостаток аденозиндезаминазы угнетает синтез Б-аденозилметионина, который является донатором метильных групп для ДНК. Деоксиаденозин- трифосфорная кислота (дАТФ) при ее накоплении в клетках тормозит активность рибонуклеотидредуктазы, без достаточного уровня которой не происходит фазы Б цикла клеточного роста. Это ведет к прекращению синтеза ДНК и включению дАТФ в полиаденилированную рибонуклеиновую кислоту (РНК), что угнетает синтез РНК и разделение ДНК на отдельные нити. В результате не происходит дифференциации клеток-предше- ственников В- и Т-лимфоцитов. Врожденный дефицит аденозиндеаминазы вызывает тяжелый комбинированный иммунодефицит у 40% больных с этим синдромом.
Иммунодефицит, обусловленный недостатком аденозиндезаминазы, чаще всего проявляет себя кандидозом полости рта, трудно устранимым поносом, задержкой роста и другими инфекциями и дефектами развития. Общее число лимфоцитов в крови находится на уровне более низком, чем 0,5*109/л. Особенно мало в крови Т-лимфоцитов. Лимфопения (патологически низкое содержание лимфоцитов в крови) приводит к эозинофилии. В моче и сыворотке крови больных повышено содержание аденозина и деоксиаденозина.
Врожденный дефицит активности нуклеозидфосфорилазы наследуется по аутосомно-кодоминантному типу. Расстройства пуринового обмена у больных с иммунодефицитом обуславливают аккумуляцию в иммуноком- петентных клетках деоксигуанозинтрифосфата, что приводит к иммунодефициту через торможение в них активности рибонуклеотидредуктазы. Вследствие низкой активности рибонуклеотидредуктазы блокируется деление клеток и преимущественно на уровне Т-клеток возникает иммунодефицит. Данный иммунодефицит приводит к частым инфекциям легких, мочевыводящих путей. Нередко иммунодефициту сопутствуют аутоиммунная гемолитическая анемия и гипоплазия клеточных элементов костного мозга. В циркулирующей крови выявляют патологически низкое содержание Т-клеток, в моче и сыворотке крови аномально высокое содержание мочевой кислоты (признак цитолиза неспособных к клональной экспансиии иммуно- компетентных клеток). Кроме того, в сыворотке крови возрастает концентрация свободных инозина и гуанозина.
Еще по теме ПАТОГЕНЕЗ НАИБОЛЕЕ ИЗУЧЕННЫХ ВРОЖДЕННЫХ ИММУНОДЕФИЦИТОВ НА УРОВНЕ ИММУНОКОМПЕТЕНТНЫХ КЛЕТОК:
- 5.2.3. Препараты из иммунокомпетентных клеток - иммуноцитомедины.
- Наиболее важные врожденные заболевания1
- 1.2.2. Одним из наиболее очевидных и заметных изменений клеток при старении живых организмов
- Патогенез врожденной ВИЧ-инфекции.
- Репрограммирование соматических клеток: возможности применения для изучения болезней нервной системы и разработки методов лечения
- Особенности течения туберкулеза, сочетанного с ВИЧ- инфекцией при разном уровне содержания СІ)4-клеток в периферической крови
- Изучение in vitro изменения синтетической активности опухолевых клеток асцитного варианта рака яичников крыс при действии ПеМП и винкристина
- Изучение in vitro синтетической активности клеток лимфы больных раком яичников при действии ПеМП, а также доксорубицина в сочетании с эпиталамином и рихлокаином
- 4.2. Изучение многомерных связей между антропометрическими, вариационнопульсомтрическими и дерматоглифическими признаками у больных мальчиков с врожденными пороками сердца без нарушения гемодинамики
- Нарушение ионного транспорта и преобразования энергии в митохондриях клеток с уменьшением синтеза АТФ как причина стационарного повышения уровня системного артериального давления.
- Комплексный подход к изучению параметров окислительной модификации белков и уровня молекул средней массы в клиникобиохимическом анализе эндогенной интоксикации.
- Комплексное использование модельной биологической системы для изучения Бе2+-индуцированной окислительной модификации белков и уровня молекул средней массы.
- Комплексное использование модельной биологической системы для изучения спонтанной окислительной модификации белков и уровня молекул средней массы.
- Тема 8. Диссеминированные процессы легких: классификация, диагностика, дифференциальная диагностика, клиника, патогенез, лечение наиболее часто встречающихся нозологий. Идиопатический фибризирующий альвеолит. Экзогенный аллергический альвеолит.
- 4.2.1. Этиология вторичных иммунодефицитов.