Кальциевая гипотеза болезни Альцгеймера
Нарушение кальциевого гомеостаза рассматривается как причина патологии БА в так называемой «кальциевой гипотезе». Для кальциевой гипотезы нехарактерно отрицание роли Ap или белка тау в прогрессии заболевания.
Существенным отличием кальциевой гипотезы является предположение о том, что нарушение кальциевого гомеостаза предшествует появлению амилоидных бляшек, нейрофибриллярных узелков и массовой гибели нейронов. Согласно данной гипотезе, в результате первичного нарушения кальциевого гомеостаза происходит образование амилоидных бляшек, которые в свою очередь оказывают влияние на кальциевые каналы, рецепторы и другие сигнальные молекулы, действуя по принципу положительной обратной связи (Berridge, 2010; Demuro et al., 2010; Mattson, 2010; Supnet, Bezprozvanny, 2010).Подтверждением этой гипотезы служат данные о том, что деполяризация мембраны и активация кальциевых каналов L-типа, аппликация к культуре нейронов кальциевых ионофоров или повышение внутриклеточной концентрации кальция в таких условиях, как ишемия, приводят к увеличению продукции Ap42 (Pierrot et al., 2004; Berridge, 2010). Более того, в исследованиях, проведенных на фибробластах людей с НБА и мышиных трансгенных моделях БА, было показано, что нарушение кальциевого гомеостаза предшествует проявлению симптомов и гистологических маркеров БА (Ito et al., 1994; Gibson et al., 1996). Существует также связь между нарушением кальциевого гомеостаза и появлением нейрофибриллярных узелков, образованных цитоскелетным белком tau. Было показано, что нейроны с высоким уровнем экспрессии кальций-зависимой протеазы кальпаин и кальмодулин-зависи- мой киназы II с наибольшей вероятностью образуют нейрофибриллярные узелки на ранних стадиях развития заболевания в мозге больных БА (McKee et al., 1990). Чрезмерная активация глутаматных рецепторов в нейронах гиппокампа приводит к изменениям в работе тау и микротрубочек и образованию структур, подобных нейрофибриллярным узелкам (Mattson, 1990).
Также на первичной культуре кортикальных нейронов было напрямую показано кальций-зависимое фосфорилирование и дефосфорилирование белка тау, опосредованное киназой GSK-3P и фосфатазой кальцинейрин (Pierrot et al., 2006).Уникальность иона кальция заключается в его участии в большом количестве клеточных процессов: возбудимости мембраны, выбросе нейромедиаторов, росте аксона, активности митохондрий, внутриклеточной передаче сигналов и экспрессии генов, клеточной дифференцировке и апоптозе. Кальций является универсальным вторичным посредником и участвует в передаче сигналов, регулируя активность многих ферментов, таких как кальмодулин-зависимые киназы, протеинкиназа С, NO-синтаза, протеаза кальпаин, фосфотаза кальцинейрин, а также транскрипционных факторов NFkB, NFAT и CREB (Bito et al., 2003; Toescu, Verkhratsky, 2007; Amici et al., 2009).
В регуляцию концентрации Ca2+ в нейронах вовлекается большое количество Ca^-каналов: потенциал-управляемые Ca2+-каналы плазматической мембраны (VGCC), NMDA рецепторы, AMPA-рецепторы, кальциевый канал CALHM1 и де- по-управляемые кальциевые каналы. Высвобождение Ca2+ из ЭР осуществляется рецепторами инозитол-1,4,5-трисфосфата (IP3R) и рианодиновыми рецепторами (RyR) (LaFerla, 2002; Thibault et al., 2007; Emptage et al., 2010; Gallego-Sandin et al.,
2011) . Помпа SERCA в ЭР, Ca^-помпа плазматической мембраны и Na+ZCa^-об- менник плазматической мембраны контролируют концентрацию Ca2+ в цитоплазме в узком диапазоне значений. В формировании цитоплазматических Са2+-сигналов важную роль играют митохондрии. Митохондриальный Ca^-переносчик (MCU) является ионным каналом, который вовлекается в мощный и быстрый вход кальция в митохондрии (LaFеrla, 2002) (рис. 1).
Внутриклеточная концентрация кальция в нейронах регулируется, в том числе за счет кальциевых буферов, основу которых представляют кальций-связывающие белки. К таким белкам относятся кальбандин, парвальбумин и кальретинин, широко экспрессирующиеся в центральной нервной системе.
Отделы головного мозга, нейроны внутри одного отдела мозга и нейроны различных нейромедиаторных систем отличаются по преобладающей форме, уровню экспрессии кальций-связы- вающего буферного белка и, соответственно, по буферной емкости. Таким образом, нейроны отличаются по чувствительности к изменению концентрации кальция (Toescu, Verkhratsky, 2007).Такое многообразие Ca2+-зависимых элементов обеспечивает возможность тонкой Ca^-зависимой регуляции нейрональных функций во временной шкале, варьирующей от микросекунд (как в случае с Сa2+-зависимым слиянием синаптичес-
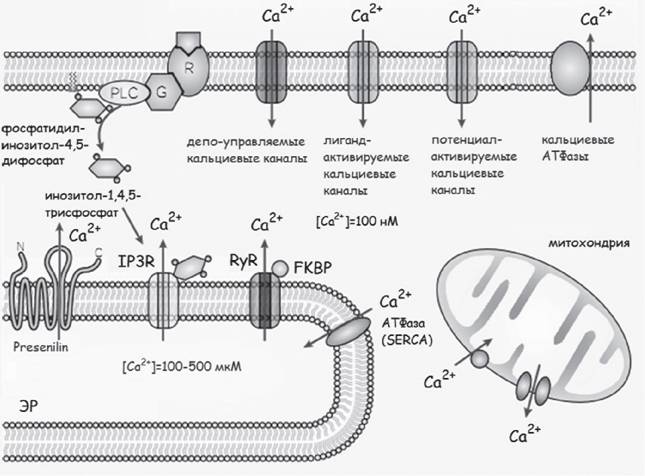
Рис. 1. Регуляция концентрации кальция в нейронах. Цитируется по (La Ferla, 2002) с изменениями. Объяснение в тексте
кого пузырька с пресинаптической мембраной) до секунд и минут (как в случае с Ca^-зависимым фосфорилированием и дефосфорилированием) и до дней и даже лет (как в случае с Ca^-зависимыми изменениями в нейрональной экспрессии генов). Эти Ca^-зависимые процессы ведут к кратковременным и долговременным изменениям возбудимости нейронов (посредством изменения активности ионных каналов и характера их экспрессии) и к изменениям синаптической передачи (посредством модификации синаптической «машинерии» и облегчения формирования или разобщения синаптических связей) (Harney et al., 2006).
Процесс запоминания связан с активацией долгосрочной потенциации и увеличением пластичности глутаматергических синапсов в гиппокампе. Долгосрочная потенциация запускается за счет достижения критической концентрации Са2+ в нейронах при активации глутаматных рецепторов AMPAR, NMDAR и mGluR в ответ на выброс глутамата из пресинаптических окончаний. Поддержание долгосрочной потенциации происходит за счет Са2+ в качестве вторичного посредника (Harney et al., 2006). Процесс стирания памяти связан с активацией долгосрочной депрессии, которая происходит при меньших концентрациях Са2+ в цитоплазме.
Основная роль в регуляции долгосрочной депрессии в гиппокампе отводится mGluR и кальций-за- висимой фосфатазе кальцинейрин. Кальцинейрин может активировать эндоцитозAMPAR и NMDAR, уменьшая, таким образом, силу синаптической связи между нейронами (Foster, 2007; Toescu, Verkhratsky, 2007).
Риск развития БА увеличивается с возрастом. Для объяснения временных изменений в мозге, способствующих развитию заболевания, была предложена интегральная модель зависимых от возраста изменений в обороте гиппокампального Ca2+ (Gant et al., 2006). Сравнительные исследования нейронов молодых и старых грызунов показали, что основные элементы Са2+-сигнализации в нейронах подвергаются значительным возрастным изменениям (Toescu, Verkhratsky, 2007) В стареющих нейронах было обнаружено увеличение концентрации Ca2+ через усиление высвобождения Ca2+ из внутриклеточных депо через IP3R и RyR, усиление входа Ca2+ через потенциал-активируемые кальциевые каналы L-типа, увеличение медленной следовой гиперполяризации вследствие активации Ca2+-зависимых K- каналов, уменьшение вклада NMDAR-опосредованного входа Ca2+, уменьшение цитоплазматической буферной емкости. Такие изменения приводят к усилению восприимчивости к индукции долгосрочной депрессии и к увеличению пороговой частоты для запуска долгосрочной потенциации в стареющих нейронах (Foster, 2007). Таким образом, возрастные изменения могут способствовать усилению тех патологических изменений в кальциевой сигнализации, которые предшествуют развитию симптомов БА.
6.
Еще по теме Кальциевая гипотеза болезни Альцгеймера:
- Патогенез болезни Альцгеймера и кальциевый гомеостаз
- Гены, ассоциированные с болезнью Альцгеймера. Наследственная болезнь Альцгеймера
- Болезнь Альцгеймера
- Сердечно-сосудистая система и болезнь Альцгеймера: клинические данные
- Болезнь Альцгеймера - клиника, диагностика и лечение
- Болезнь Альцгеймера
- Эволюция представлений о диагностике и систематике болезни Альцгеймера
- Механизмы болезни Альцгеймера
- Нарушение активности мозга на модели болезни Альцгеймера
- 1.7.3. Болезнь Альцгеймера
- Роль амилоида-р в болезни Альцгеймера
- Активация компенсаторных механизмов при болезни Альцгеймера