Гипотеза нейроиммуномодуляции
Представления, сложившиеся в рамках амилоидной и тау-гипотез, не могли объяснить весь фактический материал, накопленный в многочисленных исследованиях. Постепенно происходило формирование более широких представлений о патогенезе БА, в которых рассматривались все патологические составляющие заболевания, что, в свою очередь, позволило определить новые терапевтические мишени.
При рассмотрении БА с позиции ее мультифакторной природы ряд исследователей придерживается мнения, что возникающая цепь сигналов о повреждении передается различным клеткам мозга (главным образом микроглии), которые, в свою очередь, запускают ответные реакции со стороны системы нейроиммуномодуляции. Предполагается, что такие патологические сигналы в конечном итоге могут приводить к самоагрегации тау-белка с образованием олигомеров и фибрилл (Maccioni et al., 2010).Воспаление - это процесс, тесно связанный с развитием целого ряда нейродегенеративных расстройств, в том числе БА. Многие существующие гипотезы объясняют патогенез БА, но не дают ясности относительно ранних событий, которые инициировали метаболические и клеточные изменения, приведшие к нейродегенеративным процессам (Rojo et al., 2008). В течение последних пяти лет появились
дополнительные свидетельства в поддержку представлений о том, что нарушения в работе системы взаимодействия между глиальными клетками и нейронами играют важную роль в процессе нейрональной дегенерации. Нейроны и клетки глии совместно с кровеносными сосудами образуют интегрированную систему функционирования мозга. Изучение патологических изменений в сигнальных связях между нейронами и глией привело к появлению гипотезы нейроиммуномодуляции (рис. 4).
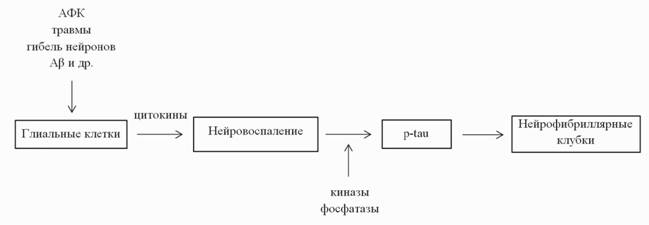
Рис. 4. Патогенез БА в рамках гипотезы нейроиммуномодуляции
Имеющиеся данные говорят о важной роли иммунной системы при старении мозга и развитии нейродегенеративных болезней, при этом взаимодействие между указанными системами определяет прогрессию патологического процесса (Lucin, Wyss-Coray, 2009).
В данном контексте микроглия и резидентные макрофаги центральной нервной системы являются ключевым фактором в регулировании локальной окружающей среды клеток и определяют статус воспалительного процесса. Длительное активирование микроглии после острого повреждения и при хронических заболеваниях говорит в пользу того, что эти клетки имеют иммунную память о тканевом повреждении и дегенерации. Фенотип микроглии также изменяется при системных инфекциях или воспалении (Perry et al., 2010). Идея о том, что изменения в иммуномодуляции критичны для патогенеза БА, представляет собой наиболее интегративный взгляд на развитие патологии, а дальнейшие исследования позволят выявить роль воспалительного процесса при данном заболевании.Исследования на культурах клеток микроглии и нейронов совместно с экспериментами на животных моделях, а также клинические данные подтверждают, что эндогенные повреждающие факторы, такие как Ap-олигомеры, активные формы кислорода, перенасыщение железом, недостаток фолата, травмы головы, определенные виды липопротеинов низкой плотности и гомоцистеин, являются триггером для активации клеток микроглии. Провоспалительные цитокины играют двойственную роль и могут как усиливать нейродегенеративный процесс, так и оказывать нейропротекторное действие. Это равновесие смещается в сторону нейродегенерации под действием целого набора выявленных факторов риска, которые запускают врожденные системы передачи сигналов о повреждении, активируют
микроглию и способствуют высвобождению провоспалительных цитокинов (Fernandez et al., 2008; Maccioni et al., 2009).
Развитие патологического процесса при БА сопровождается накоплением повреждений паренхимы мозга, которые предшествуют манифестации клинических симптомов. Считается, что нарушения в функционировании ткани инициируют систему сигналов о повреждении и запускают нейровоспалительный процесс. Такие сигналы через Toll-подобные рецепторы, рецептор высокогликозилированных конечных продуктов (RAGE) или другие глиальные рецепторы активируют иммунную систему, что в свою очередь приводит к аномальному высвобождению цитокинов и запуску каскада нейродегенерации.
Описанные события сопровождаются активацией NFkB с последующим высвобождением цитокинов, таких как TNF-a, IL-6, IL-ip. Сверхпродукция данных медиаторов может приводить к активации киназ GSK3P, cdk5, а также ингибированию фосфатаз, например PP1. В результате происходит накопление гиперфосфорилированной формы белка тау и его агрегация в нейротоксичные олигомерные формы. Агрегация тау-белка - это одно из конечных ключевых событий в патогенезе БА (Farias et al., 2011; Morales et al., 2010).Данные, свидетельствующие о связи нейровоспалительных процессов и гиперфосфорилирования тау, были получены в исследованиях маркеров нейропатологии и на трансгенных мышиных моделях БА. Активированная микроглия обнаруживается post mortem в аутопсийном материале больных с различными тау-патиями, включая БА и ФТД (Gebicke-Haerter, 2001). У трансгенных мышей линий 3xTg и rTg4510, моделирующих важные аспекты БА, введение липополисахаридов, вызывающих системное воспаление, усиливает гиперфосфорилирование тау (Kitazawa et al., 2005; Lee et al., 2010а). У мышей, моделирующих тау-патологию, активация микроглии предшествует формированию нейрофибриллярных клубков. К тому же, иммуносупрессия с помощью FK506 у молодых мышей линии P301S ослабляет развитие тау-патологии и увеличивает среднею продолжительность жизни модельных животных (Yoshiyama et al., 2007). Таким образом, можно сделать заключение, что нейровоспалительная реакция связана с нарушениями в функционировании тау белка. В то же время, было показано, что введение липополисахарида не влияет на развитие церебрального амилоидоза у модельных мышей (Kitazawa et al., 2005), а по данным некоторых авторов, в используемых ими экспериментальных системах даже способствует снижению количества амилоидных отложений (DiCarlo et al., 2001; Herber et al., 2007; Herber et al., 2004; Quinn et al., 2003). На крысиной модели тау-патии, где причиной развития нейродегенеративного процесса служит укороченная форма тау-белка человека, показана индукция воспалительного ответа, проявляющегося в повышении экспрессии «молекул иммунитета» (CD11a,b, CD18, CD4, CD45, CD48), морфологической активации клеток микроглии и инфильтрации лейкоцитов (Zilka et al., 2009).
С другой стороны, медиаторы воспаления, такие как интерлейкин-1 (IL-1), интерлейкин-6 (IL-6) и оксид азота, высвобождающиеся из астроцитов, могут усиливать фосфорилирование тау и формирование нейрофибриллярных клубков (NFTs) in vitro (Li Y. et al., 2003; Quintanilla R.A. et al., 2004; Saez T.E. et al., 2004). В ряде работ показано, что избыточное фосфорилирование тау, вызванное высвобождением интерлейкинов, вероятно, опосредовано через сигнальные пути p38-MAPK и CDK5/p35. Кроме того, активация микроглии и воздействие микроглиального провоспалительного цитокина TNFa стимулирует образование активных форм кислорода, что в свою очередь индуцирует накопление склонного к агрегации тау в нейритах (Gorlovoy et al., 2009).
Было обнаружено, что рецептор к фракталкину (CX3CR1), принимающий участие в коммуникации между клетками микроглии и нейронами, играет ключевую роль в развитии когнитивных нарушений при БА. У трансгенных мышей с мутантным геном белка тау человека и нокаутных по гену Cx3cr1, наблюдается усиление тау-патологии, которое коррелирует с повышенным уровнем активности p38-MAPK. Эксперименты in vitro показывают, что активация микроглии повышает уровень активной p38-MAPK и усиливает гиперфосфорилирование тау в нейронах, и эти эффекты могут быть заблокированы антагонистами рецептора к IL-1 и специфическими ингибиторами p38-MAPK. Эти данные указывают на то, что CX3CR1 и ^-1/р38-МАРК-пути могут служить новой терапевтической мишенью для разработки терапии тау-патий (Bhaskar et al., 2010). Интересным представляется тот факт, что недостаточность CX3CR1 способна ослаблять Ap-патологию, препятствуя гибели нейронов кортикального слоя II в мозгу 3xTg-мышей (Fuhrmann et al., 2010). Исследования на моделях церебрального амилоидоза без тау-патологии показали, что блокировка сигнального пути CX3CL1-CX3CR1 (лиганд-рецептор) действительно снижает накопление Ap, оказывая влияние на микроглиальный фагоцитоз внеклеточных Ap-агрегатов (Lee et al., 2010b; Liu et al., 2010). Таким образом, недостаточность CX3CR1 оказывает разнонаправленное действие на две основных патологических составляющих БА: формирование внеклеточных амилоидных отложений и образование внутриклеточных агрегатов тау-белка (Bhaskar et al., 2010).
В данных работах не была проведена оценка функционального состояния животных, в то время как в работе С. Чо с сотрудниками на модели hAPP-J20 было показано, что при удалении CX3CR1 вместе с уменьшением количества амилоидных отложений наблюдается нарушение когнитивной функции, что выявляется при анализе памяти в тестах «пассивное избегание» и «распознавание нового объекта» (Cho et al., 2011).Еще одним подтверждением гипотезы нейроиммуномодуляции являются эпидемиологические данные, показывающие, что у пациентов, хронически принимающих нестероидные противовоспалительные средства, снижен риск БА (McGeer et al., 2006) и наблюдается значительно менее выраженная манифестация БА. Таким образом, ослабление воспалительного процесса в мозге способствует замедлению развития заболевания (McGeer et al., 1996). Однако проведенные клинические испытания основных нестероидных противовоспалительных средств не показали выраженного эффекта при лечении БА (Rojo et al., 2008). Генетические и эпидемиологические исследования показали, что повышенная выработка TNFa является фактором риска для БА. У пациентов с БА обнаруживается избыток TNFa в цереброспинальной жидкости, а положительный эффект ингибитора TNFa - препарата этанерцепта - указывает на обратимый характер TNFa-зависимого патофизиологического механизма БА и схожих заболеваний (Tobinick, 2010).
4.
Еще по теме Гипотеза нейроиммуномодуляции:
- Источники и виды Y гипотез
- 3. Гипотеза
- Выдвижение гипотез
- 10. Уровни гипотез, проверяемых в психологическом эксперименте.
- 5. Требования к формулировкам причинно-следственных гипотез.
- Основные гипотезы патогенеза БА
- Кальциевая гипотеза болезни Альцгеймера
- 2.4 Построение диагностической гипотезы
- 39. Специфика гипотез проявляемых в факторном эксперименте.
- 3. Понятие об эмпирической проверки научных гипотез.
- Д. Опровергнутые гипотезы.
- Проверка гипотез
- Гипотезы о происхождении тормозящего действия условного раздражения.
- Гипотеза о распределенности энграммы