АТЕРОСКЛЕРОЗ
Атеросклероз - отложение в интиме сосудов атерогенных липопроте- инов низкой плотности (ЛПНП) вследствие взаимодействие гладкомышечных клеток стенок сосудов с атерогенными липопротеинами при их высокой концентрации в циркулирующей крови.
Атеросклеротическое перерождение сосудов во многом определяет пролиферация миоцитов сосудистой стенки, индуцируемая цитокинами тромбоцитов, активированных мононук- леаров, а также агентами аутокринной регуляции (митогенами) самих гладкомышечных клеток. Термин атеросклероз описывает изменения сосудистой стенки при этом системном типовом патологическом процессе. Слово «атере» (греч.) означает «кашица» и указывает на образование в стенках сосудов кашицеобразных липидных отложений с их дальнейшим склерозированием и уплотнением.По определению Всемирной организации здравоохранения «Атеросклероз - это вариабельная комбинация изменений внутренней оболочки (интимы) артерий, включающая накопление липидов, сложных углеводов, фиброзной ткани, компонентов крови, кальцификацию и сопутствующие изменения средней оболочки (медии)».
При гистологическом исследовании пораженных атеросклерозом участков сосудистой стенки выявляют характерное патологическое образование, атеросклеротическую бляшку (рис. 11.1). Ее формируют липиды, лейкоциты, гладкомышечные клетки и межклеточное вещество интимы артерий. Образованию бляшки предшествует адгезия (прилипание) циркулирующих моноцитов и лимфоцитов к артериальному эндотелию с последующим локальным накоплением пенистых клеток. Пенистые клетки представляют собой активированные мононуклеары, видоизмененные в результате интенсивного эндоцитоза липидов.
До сих пор общественное сознание связывает атеросклероз с пропитыванием холестерином сосудистой стенки, что верно лишь отчасти. Холестерин и триглицериды переносятся во внеклеточном пространстве липопротеинами. Не гиперхолестеринемия (рост концентрации холестерина выше верхнего предела нормальных колебаний) ведет к атероскдерозу, а аккумуляция в сосудистой стенке определенных липопротеинов.
Некоторые ли- попротеины можно считать атерогенными, другие неатерогенными, а третьи антиатерогенными.Под атерогенными следует понимать липопротеины, которые проникают в сосудистую стенку, где происходит их эндоцитоз макрофагами, которые в результате эндоцитоза превращаются в пенистые клетки. В последующие стадии патологического процесса происходит отложение аморфного холестерина и его кристаллов в межклеточных пространствах сосудистой стенки, то есть вне пенистых клеток. При гиперли- пидемии второго типа, которая часто приводит к атеросклерозу, пенистые клетки, насыщенные липидами, находят во внутренней оболочке сосудистой стенки.
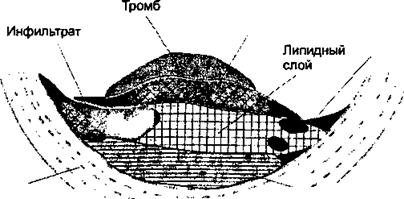 |
| Средняя оболочка чч кровеносных сосудов |
| Фибрин |
| Эндотелиолит |
| ^ Пролиферирующие гл адкомышеч ные клетки |
| Рис. 11.1. Схематическое изображение строения атеросклеротической бляшки, вызывающей стенокардию |
Поверхность макрофагов содержит рецепторы к липопротеинам очень низкой плотности и непостоянные рецепторы к измененным в результате окисления липопротеинам низкой плотности («рецепторы-мусорщики»). Предположительно активация мононуклеаров через связывание этих рецепторов с окисленными атерогенными липопротеинами или липопротеинами очень низкой плотности ведет к активации макрофагов, в результате которой они приобретают способность к эндоцитозу атерогенных липопротеинов.
В ранней стадии атеросклеротического поражения сосудистой стенки в ней выявляют липидные прожилки и пенистые клетки. Этот начальный этап формирования атеросклеротической бляшки вызывает активация непостоянно присутствующих на клеточной поверхности макрофагов рецепторов к окисленным липопротеинам низкой плотности (ацетил-ЛПНП- рецепторы, «рецепторы-мусорщики»).
Мусорщиками эти рецепторы называют потому, что они обладают высоким сродством к «мусору» в виде окисленных ЛПНП. Изменения ЛПНП в результате окисления, которые обуславливают их особо высокое сродство к рецепторам-мусорщикам включают:♦ трансформацию лецитина в составе липопротеинов в лизолецитин;
♦ окисление холестерина;
♦ рост отрицательного заряда и плотности липопротеинов;
♦ снижение содержания в ЛПНП полиненасьпценных жирных кислот.
Распад структурного апопротеина липопротеинов низкой плотности В-
100 с высвобождением гистидина, лизина и пролина. Окисленные липоп- ротеины служат хемоаттрактантами и активаторами для макрофагов сосудистой стенки, одновременно являясь для них и объектом эндоцитоза.
Как результат патогенных межклеточных взаимодействий атеросклероз представляет собой патологическое изменение стенок артерий большого и среднего диаметра, которые составляют и вызывают локальная аккумуляция в интиме липидов и макрофагов, миграция и пролиферация шадкомышеч- ных клеток, а также отложения вещества внеклеточного матрикса. Патогенные межклеточные взаимодействия, приводящие к атеросклерозу, реализуются не только через действие факторов роста, но и в результате эффектов цитокинов флогогенов, медиаторов воспаления и экспрессии адгезивных молекул. Начальным этапом патогенеза атеросклероза как последовательности межклеточных взаимодействий является адгезия моноцита циркулирующей крови к эндотелиальным клеткам с их последующей миграцией в интиму. При этом индукторы атеросклероза окисленные (модифицированные) липоп- ротеины низкой плотности, воздействуя на лейкоциты циркулирующей крови и эндотелиальные клетки, вызывают экспрессию на их поверхности адгезивных молекул. Так, лизофосфатидилхолин - элемент молекулы окисленных атерогенных липопротеинов - индуцирует экспрессию межклеточной адгезивной молекулы-1 на поверхности эндотелиальных клеток. Известно, что атерогенные липопротеины оказывают на эндотелий прямые и непрямые влияния, повышающие экспрессию на их поверхности эндотелиально-лейкоцитарных адгезивных молекул (ЭЛАМ).
Непрямой эффект атерогенных липопротеинов на рост экспрессии ЭЛАМ состоит в стимуляции паракринной секреции гладкомышечными клетками и мононуклеарными макрофагами сосудистой стенки. Адгезия лейкоцитов к сосудистой стенке служит первым этапом их проникновения в интиму. Там активированные моноциты секрети- руют ряд активаторов эндотелия и хемоаттрактантов, стимулирующих дальнейшую инфильтрацию очага атеросклеротического повреждения моноцитами и лимфоцитами. Патогенное функционирование иммунокомпетентных клеток в качестве эффекторов атеросклероза указывает на участие в развитии атеросклероза иммунопатологической реакции.Атеросклероз во многом представляет собой хроническое воспаление сосудистой стенки, протекающее с преобладанием пролиферативного компонента, основными клеточными эффекторами которого являются моноциты циркулирующей крови, мононуклеарные фагоциты субинтимального слоя, гладкомышечные сосудистые клетки, активированные атерогенны- ми липопротеинами или в результате межклеточных взаимодействий.
Воспаление в очаге атеросклеротического поражения усиливает взаимосвязанное с ним локальное свертывание крови, которое могут вызвать сами атерогенные липопротеины. Так, атерогенный липопротеин(а), содержит гликопротеин(а), связанный с апопротеином В. Идентичность структуры липопротеина(а) строению плазминогена может обуславливать внутри- сосудистый тромбогенез через конкурентное связывание рецепторов к плазминогену на поверхности эндотелиоцитов.
Таков патогенез индукции атеросклероза через образование прожилок атерогенных липопротеинов и пенистых макрофагов в сосудистой стенке как начального этапа образования атеросклеротической бляшки. В нем особую роль как эффекторы и регуляторы патологического процесса играют макрофаги сосудистой стенки, которые сами могут индуцировать окисление ЛПНП через высвобождение свободных кислородных радикалов и (или) активацию липооксигеназы. Так как макрофаги представляют собой эффектор единой системы иммунитета организма, то мы вправе предположить роль в развитии атеросклероза нервного эндогенного этиологического фактора, столь сильно меняющего состояние системы иммунитета, и ее макрофа- гального звена в частности.
Кроме того, патогенный стресс как состояние, которое характеризует системная интенсификация свободнорадикального окисления липидов, может повышать уровень окисления ЛПНП и на уровне сосудистой стенки предрасполагать к атеросклерозу.Пенистые клетки высвобождают ряд цитокинов, чье действие вызывает пролиферацию клеточных элементов, и в особенности миоцитов гладкомышечных элементов сосудистой стенки. Кроме того, цитокины активированных макрофагов активируют эндотелиоцигы, что ведет к росту экспрессии их тромбогенного потенциала. Цитокины пенистых клеток активируют и нейтрофилы циркулирующей крови, что вызывает воспаление с полимор- фонуклеарами в качестве его клеточных эффекторов. В результате в составе атеросклеротической бляшки находят пролиферирующие миоциты сосудистой стенки, агрегаты активированных тромбоцитов, других форменных элементов крови, активированные нейтрофилы и нити фибрина. Все это характеризует атеросклеротическую бляшку как очаг воспаления и локус тромбоза.
Макрофаги, окисленные липопротеины, ацетил-ЛПНП-рецепторы играют определяющую роль в индукции атеросклероза. В дальнейшем процесс образования атеросклеротической бляшки теряет связь с этими этиологическими факторами, то есть через патогенные межклеточные взаимодействия происходит его эндогенизация.
Одно из наиболее впечатляющих достижений медицинской генетики последних десятилетий - выяснение генетической детерминированности гиперхолестеринемии и высокой концентрации в плазме крови липопроте- инов низкой плотности как причины атеросклероза. Исследовав 500 больных, выживших после инфаркта миокарда, Гольдштейн, Браун и Мотульс- кий более, чем в половине случаев выявили семейную гиперхолестеринемию, семейную гиперлипидемию или комбинированную гиперлипидемию. Было установлено, что эти расстройства липидного обмена представляли собой патогенные фенотипические признаки, детерминированные одним геном, то есть были моногенными наследственными расстройствами липидного обмена. Последующие исследования клеточных культур фибробластов кожи, лимфоцитов, и миоцитов аортальной стенки, взятых у гомозиготных и гетерозиготных по данному гену больных, позволили выявить генетически детерминированные дефекты связывания молекулярного комплекса липопротеины низкой плотности-холестерин с наружной клеточной мембраной и его последующего пиноцитоза.
Взаимодействие молекулярного комплекса холестерин-ЛПНП при нормальной экспрессии ЛПНП-рецептора на поверхности клеток ведет к пино- цитозу молекулярного комплекса. После пиноцитоза комплекс инкорпорируется в лизосомы, где и происходит высвобождение свободного холестерина. Рост концентрации свободного холестерина в клетке снижает активность ключевого фермента внутриклеточного синтеза холестерина гид- роксиметилппотарил-коэнзим А-редуктазы. Наследуемая по аутосомально-
доминантному типу недостаточность ЛПНП-рецепторов ведет к снижению пиноцитоза комплекса холестерин-ЛПНП и к падению концентрации свободного холестерина в клетках. В результате низкого содержания свободного холестерина в клетках с низким содержанием на наружной поверхности ЛПНП-рецепторов в них высока активность гидроксиметилглютарил-коэн- зим А-редуктазы. Это ведет к интенсивному образованию холестерина клетками, его высвобождению во внеклеточное пространство и росту в нем содержания атерогенных липопротеинов переносчиков холестерина. Гиперхолестеринемия вызывает атеросклероз не через пропитывание стенки сосудов холестерином, а через повышение интенсивности образования печенью и высвобождения ею в кровь атерогенных липопротеинов (схема 11.3). Кроме того, гиперхолестеринемию при семейной гиперхолестери- немии и других гиперлипидемиях, связанных с атеросклерозом, обуславливает и низкий уровень связывания комплекса ЛПНП-холестерин с ЛПНП-рецепторами наружных клеточных мембран. Гиперхолестеринемия алиментарного генеза также повышает риск атеросклероза через увеличение в крови концентрации ЛПНП и других атерогенных липопротеинов, переносящих холестерин во внеклеточном пространстве. Рост концентрации ЛПНП повышает массу циркулирующих с кровью продуктов их окисления, вступающих во взаимодействие с рецепторами-мусорщиками наружных клеточных мембран макрофагов, то есть повышает вероятность реализации инициирующего момента атеросклероза.
Еще по теме АТЕРОСКЛЕРОЗ:
- Атеросклероз
- АТЕРОСКЛЕРОЗ
- АТЕРОСКЛЕРОЗ
- Этиология атеросклероза:
- 1.7.1. Атеросклероз
- Атеросклероз
- Атеросклероз
- Клинические проявления атеросклероза:
- Тема занятия. АТЕРОСКЛЕРОЗ И АРТЕРИОСКЛЕРОЗ. ИШЕМИЧЕСКАЯ БОЛЕЗНЬ СЕРДЦА
- Роль нарушений липидного обмена в патогенезе атеросклероза
- Патогенез атеросклероза: