Амилоид-деградирующие ферменты
Несмотря на то что накопление Ap в ткани мозга длительное время считалось необратимым процессом, сравнительно недавно было обнаружено существование эндогенных биохимических путей его деградации и удаления из ткани мозга (для обзора см.
Nalivaeva et al., 2012c). Как было показано рядом авторов, некоторые хорошо известные протеазы мозга способны гидролизовать Ap в физиологических условиях. Это привело к пересмотру имеющихся представлений о метаболизме амилоидного пептида. К числу основных амилоид-деградирующих ферментов сейчас относят неприлизин (НЕП), эндотелин-конвертирующий фермент (ЕСЕ-1), инсулин-деградирующий фермент (ИДФ; инсулизин) и плазмин, которые способны расщеплять Ap как in vitro, так и in vivo (Nalivaeva et al., 2008, 2012а,Ь,с). На рис. 4 схематично показаны участки молекулы Ap, на которых возможно его протеолитическое расщепление различными ферментами. Снижение уровня экспрессии или активности этих ферментов при патологических условиях может вести к накоплению Ap и развитию БА или других нейродегенеративных патологий. С другой стороны, повышение экспрессии и активности амилоид-деградирующих ферментов как фармакологическими средствами, так и посредством генной терапии открывает новое направление в профилактике и терапии Ap-опосредованной нейродегенерации и БА.Неприлизин. Неприлизин (НЕП, нейтральная эндопептидаза-24.11) является цинкзависимой металлопептидазой, впервые описанной в ткани почек как фермент, деградирующий пептидные гормоны (см. обзор Turner, Tanzawa, 1997). Этот фермент также известен как энкефалиназа благодаря его способности расщеплять
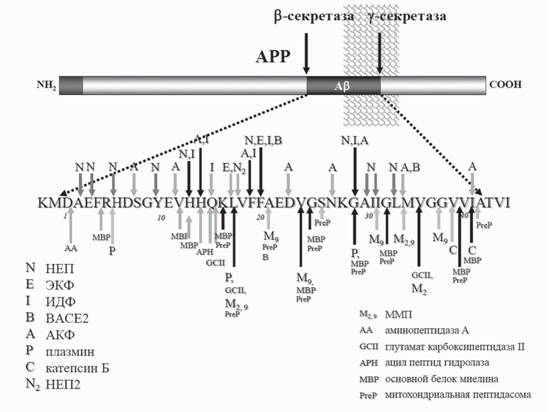
Рис. 4. Участки расщепления Ар пептида амилоид-деградирующими ферментами Ap образуется из АPP путем последовательного действия P- и у-секретаз. Ар пептиды, состоящие, как правило, из 40 или 42 аминокислотных остатков, расщепляются неприлизином (Н), эндоте- лин-конвертирующим ферментом (Е), инсулин-деградирующим ферментом (И), матричными металлопептидазами MMP-2, 3, 6 или 9 (M), плазмином (П), митохондриальной пептидазой PreP, а также рядом других ферментов, которые перечислены на рисунке
энкефалин и прерывать пептидергическую нейропередачу.
НЕП представляет собой мембраносвязанный гликозилированный эктофермент с молекулярным весом 93 кДа, активный центр которого обращен во внеклеточное пространство. НЕП способен расщеплять пептидные связи внутри молекул низкомолекулярных пептидов по гидрофобным аминокислотным остаткам (Erdos, Skidgel, 1989). НЕП обладает довольно широкой субстратной специфичностью и регуляторной активностью. Благодаря этим свойствам НЕП принимает участие в регуляции разнообразных физиологических процессов, включая сердечно-сосудистую деятельность, воспалительные процессы, миграцию и пролиферацию клеток, а также развитие опухолей (Usmani et al., 2002; Nalivaeva et al., 2012c). Ключевая роль НЕП в деградации нейропептидов подтверждает возможную физиологическую роль Ap, предложенную А.Р. Кудиновым и TT Березовым (Koudinov, Berezov, 2004), поскольку Ap также является одним из физиологических субстратов НЕП. НЕП присутствует в головном мозге в относительно низких количествах. Так, например, если в ткани почек его содержание составляет 4% от общего содержания всех белков, то в ткани мозга его в 1000 раз меньше.Способность НЕП расщеплять Ap-пептиды делает его важным участником на арене исследований патогенеза БА (Iwata et al., 2004). Поскольку НЕП способен
расщеплять не только мономеры, но также олигомерные формы Ap и Ap (Kanemitsu et al., 2003), это его свойство является особенно важным для предотвращения образования более токсичных олигомерных форм Ap пептида. В предварительных клинических исследованиях было показано, что содержание НЕП и его мРНК у пациентов с БА является достоверно более низким, чем у контрольных пациентов соответствующего возраста (Yasojima et al., 2001). В предпринятом недавно в Японии эпидемиологическом исследовании было показано существование корреляции между уровнем экспрессии гена НЕП и развитием старческой формы БА (Sakai et al., 2004a), однако другие исследователи не смогли выявить наличие такой корреляции (Lilius et al., 2003).
Неприлизин 2. Относительно недавно был обнаружен гомолог НЕП, названный НЕП2 (также известный как NL1 и SEP), экспрессия которого в головном мозге свойственна, в основном, развивающимся и дифференцирующимся областям ЦНС и комплиментарна распределению НЕП, присутствуя в ограниченных популяциях нейронов, в спинном мозге, гипофизе и сосудистом сплетении мозга (Facchinetti et al., 2003).У мышей НЕП2 также обнаружен в большинстве перефери- ческих органов, хотя преимущественным местом его локализации является тестис (Ghaddar et al., 2000).
Важно отметить, что НЕП2 человека и грызунов (мышей и крыс) обладают разными паттернами экспрессии, ферментативными свойствами и чувствительностью к ингибиторам, что необходимо учитывать при экстраполяции данных, полученных на трансгенных мышах, для оценки функциональной роли этого фермента в ткани мозга человека (Huang et al., 2008; Whyteside, Turner, 2008). Анализ субстратной специфичности НЕП и НЕП2 человека показал, что они оба интенсивно расщепляют Ар и НЕП2 обладает большим сродством к этому субстрату (Whyteside, Turner, 2008). Принимая во внимание характер распределения НЕП2 и его ферментативные особенности, предполагают, что он является важным амилоид-деградирующим ферментом и расширяет диапазон возможности клеток и тканей катаболизировать Ap (Marr, Spencer, 2010). Недавние клинические исследования также выявили изменение уровня экспрессии НЕП2 в разных участках мозга при мягком когнитивном снижении, что позволяет рассматривать его в качестве одного из маркеров доклинической стадии БА (Huang et al., 2012).Эндотелин-конвертирующий фермент. Другим кандидатом на роль ами- лоид-деградирующего фермента является эндотелин-конвертирующий фермент (endothelin-converting enzyme, ЕСЕ-1, ЭКФ). ECE-1 является мембраносвязанным белком, на 37% гомологичным по аминокислотной последовательности НЕП (в случае сравнения белков крысы). ЕСЕ-1 также является цинк-зависимой металлопротеазой и его основная физиологическая роль состоит в превращении большого эндотелина, в мощный вазоконстриктор эндотелин-1 (Xu et al., 1994).
ЕСЕ-1 был впервые изолирован из легких крыс, а затем обнаружен в большом числе органов и тканей (Takahashi et al., 1995). Наиболее обогащены этим ферментом эндотелиальные клетки, но он также экспрессируется в экзокринных клетках, мышцах, нейронах и глии (Barnes et al., 1997). В гладкомышечных клетках ЕСЕ-1 находится в комплексе с а-актиновыми филаментами. Сравнивая ЕСЕ-1 гетерозиготных и нокаутных мышей, Е.А. Экман и коллеги показали, что концентрация Ap- пептидов в их мозге существенно выше, чем у контрольных животных, и имеет ген-зависимый характер (Eckman et al., 2003).
Как было показано этими авторами, растворимый рекомбинантный ЕСЕ-1 в условиях in vitro обладает способностью гидролизовать синтетические AP40 и AP42 на множественных участках (рис. 4).Существует еще один белок, подобный ЕСЕ, который кодируется специфическим геном и носит название ЕСЕ-2 (Emoto, Yanagisawa, 1995). ECE-2, в основном, локализован в мозге и представлен незначительными количествами в эндотелиальных и гладкомышечных клетках. ЕСЕ-2 имеет 59% идентичности с ECE-1, но отличается от него кислотным pH-оптимумом, будучи практически неактивным при нейтральных рН. Он локализован в секреторных компартментах клеток где, вероятно, принимает участие в деградации белков. ECE-2, как и ЕСЕ-1, может ка- таболизировать Ap, хотя оба этих фермента расщепляют разные его пулы - секре- тируемый и цитоплазматический (Pacheco-Quinto, Eckman, 2013).
Инсулин-деградирующий фермент. Еще одним ферментом, способным расщеплять Ар, является цинк-зависимая металлопептидаза, инсулин-деградирующий фермент (ИДФ, инсулизин). ИДФ, в основном, локализован в цитозоле, но также обнаруживается в пероксисомах (Duckworth et al., 1998). Небольшие количества этого фермента находят в плазматических мембранах, а также во внеклеточной среде, хотя механизм секреции ИДФ пока не известен. Совсем недавно появились данные о том, что продукт альтернативной транскрипции ИДФ генерирует митохондриальную изоформу этого фермента, которая способна расщеплять некоторые митохондриальные белки (Leissring et al., 2004). ИДФ расщепляет субстраты предпочтительно по гидрофобным и основным остаткам аминокислот, однако этот процесс не является строго последовательность-специфичным (см. рис. 4). Он обладает способностью расщеплять in vitro широкий спектр физиологических субстратов и имеет другие физиологические функции помимо инсулинового метаболизма. Было высказано предположение, что специфичность ИДФ обуславливается конформацией субстрата и его размерами с предпочтением к более крупным молекулам (Duckworth et al., 1998).
Как было показано, ИДФ обладает специфичностью к ращеплению Ар, ассоциированного с семейными мутациями гена АРР (Morelli et al., 2005).Роль ИДФ в катаболизме Ap была впервые предположена И.В. Курочкиным и С. Гото (Kurochkin, Goto, 1994) и исследовалась далее Д.Дж. Селко и коллегами (Qui et al., 1998). Принимая во внимание внутриклеточную локализацию ИДФ, было высказано предположение, что он может быть вовлечен в деградацию внутриклеточного Ap. Гипотеза о том, что снижение активности ИДФ может вести к развитию БА, была поддержана работами А. Перез и коллег (Perez et al., 2000). Недавно выведенная линия трансгенных мышей, моделирующих БА (McGill-Thyl- APP), и характеризуемая значительным снижением экспрессии ИДФ на ранней преклинической стадии развития патологии, рассматривается сейчас как важный инструмент в изучении механизмов регуляции и восстановления активности этого фермента (Ferretti et al., 2011).
В сравнительном исследовании ими было показано существенное снижение активности ИДФ в мозге пациентов, страдающих БА, по сравнению с контрольными пациентами того же возраста. Напротив, повышенная экспрессия ИДФ, как и
НЕП, в нейронах мозга трансгенных мышей приводила к существенному снижению уровня секретируемого ими Ap, препятствовала формированию амилоидных депозитов и связанной с ними цитопатологии, а также предотвращала преждевременную гибель АРР-мутантных животных (Leissring et al., 2003). Тем не менее, интракраниальные инъекции адено-ассоциированных векторов (AAV), экспрессирующих НЕП или ИДФ трансгенным мышам, показали, что только НЕП-содержащий вектор действительно приводил к снижению амилоидной патологии (Carty et al., 2013), что снижает потенциал возможного применения ИДФ-векторов для терапии БА.
Помимо Ар ИДФ также обладает способностью гидролизовать цитоплазматический фрагмент АРР - AICD (Edbauer et al., 2002). Повышение экспрессии ИДФ в клетках как экстраневрального так и нейронального происхождения приводило к значительному снижению в них AICD.
Тем не менее, при полном ингибировании ИДФ не наблюдалось существенного повышения уровня внутриклеточного AICD, что свидетельствует о наличии альтернативных путей его деградации.В литературе имеются данные, связывающие патогенез БА с действием и патологией метаболизма инсулина (Gasparini, Xu, 2003), в связи с чем БА часто называют дабетом 3 типа (Kroner, 2009). Предполагается, что инсулин и инсулиноподобные факторы, которые играют определенную роль в обучении и памяти, также могут быть вовлечены в развитие патологических процессов, ведущих к БА (Dore et al., 1997). Более того, нарушение метаболизма глюкозы также является одним из характерных признаков БА (Bigl et al., 2003). Как было показано, экспрессия инсулиновых рецепторов повышена в мозге больных, страдающих БА, но имеет нарушенный сигнальный механизм (Hoyer, 2004). В опытах по рецепторному связыванию было обнаружено, что Ap и Ap снижают способность инсулина связываться со своими рецепторами, а также блокируют автофосфорилирование рецепторов. Напротив, обращенный пептид Ap не изменяет связывание инсулина с рецепторами и их автофосфорилирование. Это позволяет предположить, что AP(1 и AP(1 являются непосредственными конкурентными ингибиторами рецепторного связывания и действия инсулина (Xie et al., 2002).
Рядом авторов предполагается наличие общих механизмов развития гиперин- сулинемии, диабета и БА. Так, с использованием генетически модифицированных линий мышей было показано, что полное отсутствие аллелей гена ИДФ (-/-) приводит более чем к 50-процентному снижению уровня катаболизма Ap как в мембранных фракциях мозга, так и в первичных нейрональных культурах, полученных из мозга этих животных. При этом в печени этих животных также наблюдается пониженное расщепление инсулина (Farris et al., 2004). ИДФ (-/-) мыши помимо увеличенного накопления в ткани мозга эндогенного Ap также демонстрировали гиперинсулинемию и нетерпимость к глюкозе, характерные для диабета второго типа. Эпидемиологические исследования также свидетельствуют в пользу того, что инсулин-независимый диабет второго типа связан с повышенным риском развития БА (Nicolls, 2004).
Генетические изменения внутри или вблизи гена ИДФ также связывают с увеличением риска и тяжести БА (Prince et al., 2003). В частности, на 10-й хромосоме был обнаружен большой участок (размером более 60 мегапар оснований), включающий ген ИДФ, который сопряжен с риском развития БА. Генетические изменения на этом участке ДНК позволили идентифицировать аллели риска развития БА с эффектом, сравнимым по своей значимости с мутацией аллели e4 аполипопротеина Е (Mahley et al., 2007). Однако генетические исследования в Японии и Франции не подтвердили наличия связи между полиморфизмом гена ИДФ и повышением вероятности возникновения как ранней, так и старческой форм БА (Sakai et al., 2004b). Напротив, другая группа исследователей (Blomqvist et al., 2004) обнаружили равновероятностное присутствие как нарушающих, так и протекторных аллелей гена ИДФ, связанных с патогенезом не только БА, но также и болезни Паркинсона.
Плазмин. Плазмин (EC 3.4.21.7) является сериновой протеиназой, образующийся из неактивного зимогена, плазминогена, в результате действия других сериновых протеаз, называемых активаторами плазминогена. Плазмин расщепляет многие компоненты внеклеточного матрикса и в плазме крови действует, в основном, как тромболитический фактор. В мозге плазминогеновая система вовлечена в многочисленные функции, такие как нейрональная пластичность, поддержание уровня нейротрофинов, функционирование холинергической системы, долгосрочная потенциация посредством деградации ламинина, обучение и память (Nakagami et al., 2000; Allard et al., 2012). При фокальной церебральной ишемии наблюдается активация плазминогеновой системы в мозге, сохраняющаяся в течение нескольких дней после инсульта, что предполагает ее участие в процессах деструкции внеклеточного матрикса, вторичных кровотечениях и отеке мозга (Pfefferkorn et al., 2000). Уровень плазмина также снижен в ткани мозга пациентов, страдающих БА, особенно в гиппокампе (Ledesma et al., 2000).
Роль плазмина в патогенезе БА пока полностью не ясна, хотя в литературе есть ряд наблюдений, свидетельствующих о способности плазмина катаболизировать амилоидный пептид (Van Nostrand, Porter, 1999). Так, было показано, что очищенный плазмин способен ращеплять Ap с физиологически значимой эффективностью, равной 1/10 скорости расщепления им фибрина. При этом масс-спектрометрический анализ показал, что плазмин гидролизует Ap в нескольких местах (см. рис. 4), а данные электронной микроскопии подтвердили, что плазмин способен расщеплять также Ap фибриллы. Более того, экзогенно добавленный к клеткам плазмин способен блокировать нейротоксичность, вызванную Ap (Tucker et al., 2000). Исследования ткани мозга трансгенных APP/PS1 мышей, у которых также отсутсвует ген ингибитора активатора плазминогена PAI-1, выявили более низкое содержание у них Ар, что указывает на участие плазмина в его катаболизме (Liu et al., 2011).
В работе И.Б. Кингстона и коллег показано, что агрегированные формы Ap in vitro способны стимулировать энзиматическую активность тканеспецифического активатора плазминогена (tPA) (Kingston et al., 1995). Образующийся в результате этого плазмин специфически расщепляет Ap таким образом, что усиливаются его свойства к образованию Р-складок и агрегации. Это, в свою очередь, ведет к дальнейшей стимуляции tPA и более усиленной продукции плазмина. Такой механизм обратной связи может играть отрицательную роль, поскольку избыточный протеолиз белков, вызванный активацией плазминогена, может вести к разрушению клеток сосудов головного мозга и кровоизлияниям (Van Nostrand, Porter, 1999).
Основным эндогенным ингибитором tPA в ткани мозга является белок нейро- серпин, синтезируемый в нейронах. При БА в мозге имеет место повышение содержания нейросерпина и снижение активности плазмина (Fabbro, Seeds, 2009). При нокауте гена нейросерпина у трансгенных hAPP-J20 мышей, моделирующих БА, наблюдалось резкое снижение содржания AP42, сопровождавшееся улучшением когнитивных функций, по сравнению с животными с активным геном нейросерпина (Fabbro et al., 2011). Однако, несмотря на многочисленные данные, свидетельствующие об участии плазминовой системы в катаболизме амилоидных отложений, другая группа исследователей не смогла выявить существенных изменений экспрессии плазминогена на уровне мРНК и белка в ткани мозга при БА (Barker et al., 2010). Но поскольку эти исследования проводились post mortem, интерпретация полученных данных должна проводиться с осторожностью. Другой группе исследователей также не удалось выявить различий в уровне содерждания t-PA, плазминогена и PAI-1 в ЦСЖ больных и здоровых людей, хотя они не отвергают роли плазминонгеновой системы в расщеплении амилоида и патогенезе БА (Martorana et al., 2012).
В составе плазматических мембран плазмин находится в ассоциации с холес- терин-обогащенными микродоменами, так называемыми липидными рафтами (Ledesma et al., 2003), которые считаются местами преимущественного образования амилоидного пептида (Cordy et al., 2003). Это также свидетельствует о наличии функциональной связи между плазмином, холестерином и метаболизмом Ap (Ehehalt et al., 2003).
Рядом авторов было показано, что агрегированные формы Ap также способны повышать уровень мРНК, кодирующих тканеспецифические активаторы плазминогена урокиназного типа (uPA) (Tucker et al., 2002), который тоже связывают с патогенезом БА. Как было показано в работах нескольких исследовательских групп, локус на хромосоме 10, связанный с фамильной формой БА, содержит в своем составе ген урокиназного активатора плазминогена (Ertekin-Taner et al., 2005). Эти авторы также продемонстрировали, что комбинация uPA и плазминогена вместе, но не по отдельности, ингибируют токсичность Ap, снижая его фибрилогенез и формирование депозитов Ap.
Поиск генетической связи между плазмином и риском развития БА пока не позволяет сделать однозначные заключения, хотя имеются данные, связывающие уровень содержания АР42 с полиморфизмом гена активатора плазминогена урокиназного типа (PLAU) на хромосоме 10 и спорадической формой БА (Riemenschneider et al., 2006). В проведенном недавно исследовании (Shibata et al., 2005), сравнивающем 14 наиболее часто встречающихся вариаций генов, кодирующих компоненты плазминовой системы, не было подтверждено, что полиморфизм этих генов связан с частотой встречаемости БА.
Фармакологическая или генетическая регуляция плазминогеновой системы пока остается одной из возможных стратегий профилактики и лечения БА, поскольку плазмин способен эффективно удалять внутриклеточные мономерные и внеклеточные фибриллярные формы Ар. Низкомолекулярные ингибиторы активатора плазминогена (PAI-1) могли бы эффективно поддерживать активность плазми- новой системы на желательно высоком уровне. Однако, поскольку плазмин также вовлечен в метаболизм фибрина, создание такой терапии следует проводить с осторожностью во избежание побочных эффектов в виде кровотечений.
Ангиотензин-конвертирующий фермент. Ангиотензин-конвертирующий (ангиотензин-превращающий фермент, АКФ, АСЕ) является ключевым компонентом ренин-ангиотензиновой системы (РАС) как в ЦНС, так и на периферии. АСЕ регулирует кровяное давление и функции сердечно-сосудистой системы посредством превращения ангиотензина I в мощный вазоконстриктор ангиотензин II и одновременно посредством инактивации вазодилятора брадикинина. Наличие связи между АСЕ и патогенезом БА было показано в генетических исследованиях, а метаанализ данных литературы с еще большей убедительностью показал существование прямой связи между РАС, развитием гипертензии, гипоксией/ишемией мозга и БА (для обзора см. Kehoe, Wilcock, 2007). Полиморфизм гена DCP на хромосоме 17q13, кодирующего АСЕ, ассоциирован с высоким риском развития БА, особенно в случае инсерций или делеций (I/D) на участке интрона 16 (Lehman et al., 2005). Кроме этого существует связь между развитием старческой формы БА и полиморфизмом на других участках гена АСЕ (Kehoe et al., 2003). В этой связи встает закономерный вопрос о том, что определяет связь между БА и АСЕ: его свойства, связанные с РАС, или способность расщеплять Ар. В отличие от НЕП и ИДФ, при БА уровень содержания и активность АСЕ в ткани мозга, в частности в гиппокампе, фронтальной коре и хвостатом ядре, значительно выше, чем в норме (Savaskan et al., 2001). Повышение активности АСЕ может непосредственно вести к когнитивным нарушениям в результате гиперпродукции ангиотензина II, который оказывает ингибирующий эффект на высвобождение ацетилхолина.
Как было показано Дж. Ху и соавторами (Hu et al., 2001), АСЕ способен расщеплять Ар, а также ингибировать формирование его агрегатов и токсичность в условиях in vitro, и эти свойства АСЕ подавляются в присутствии его специфического ингибитора лизиноприла.
Амилоид-деградирующие свойства АСЕ были подтверждены в работе (Hemming, Selkoe, 2005), где было показано, что АСЕ расщепляет как АР1-40, так и АРЬ
и что ингибиторы АСЕ приводят к накоплению Ар в APP-экспрессирующих клетках CHO. Более того, как оказалось, АСЕ способен превращать токсический АР1-42 в менее патогенный АР1-40 (Zou et al., 2007). Введение АСЕ ингибитора кап- топрила трансгенным мышам Tg2576, используемым как модель БА, приводило к ускоренному накоплению в ткани мозга Ар, в частности его более патогенной формы АР1 Эти данные свидетельствуют, что умеренное повышение активнос
ти АСЕ может представлять терапевтическую ценность для терапии нейродегенерации и БА. С другой стороны, вызывает опасение, что длительное применение ингибиторов АСЕ для лечения гипертензии может повышать риск развития БА. В развитие этого предположения был проведен анализ изменений уровня Ар на периферии и образования амилоидных отложений в ткани мозга, при использовании других ингибиторов АСЕ, которое не подтвердило это опасение (Hemming et al., 2007). Клиническое исследование большой группы пациентов в Японии (Ohrui et al., 2004) показало, что применение АСЕ-ингибиторов, способных проникать через гематоэнцефалический барьер (например каптоприл) и ингибировать АСЕ в ткани мозга, коррелирует с более низким процентом развития БА, чем при применении ингибиторов, которые не способны ингибировать мозговую форму АСЕ (например лизиноприл). В связи с этим высказывается предположение о существовании связи между активностью АСЕ и развитием церебральной амилоидной ангиопатии, поскольку при этой патологии показано повышение уровня содержания и активности АСЕ, вероятнее всего нейронального происхождения, в периваскулярных районах мозга (Miners et al., 2008).
В литературе имеются убедительные данные о наличии генетической связи между полиморфизмом АСЕ и развитием спорадической формы БА (Katzov et al., 2004; Bertram, Tanzi, 2008). Более того, была показана связь между полиморфизмом этого гена и развитием мягкого когнитивного снижения амнестического типа (Zhang et al., 2012).
Матричные металлопротеиназы. Матричные металлопротеиназы (ММП) представляют собой еще один класс ферментов, которые в условиях in vivo могут катаболизировать не только АРР, но и Ар (см. рис. 4 ) (для обзора см. Kauwe et al., 2007). Так, было показано, что АР140 является субстратом ММП-2 (желатиназы А), ММП-3 (стромелизина), ММП-6 и ММП-9 (желатиназы Б). Более того, Ар способен повышать уровень экспрессии этих ферментов (Roher et al., 1994; Yoshiyama et al., 2000). Среди перечисленных ферментов ММП-9 является наиболее изученной в отношении специфичности к Ар (Backstrom et al., 1996). Эти авторы показали связь между характером распределения ММП-9 и уровнем содержания Ap и обнаружили присутствие этого фермента в непосредственной близости от амилоидных бляшек. ММП-9 in vitro способна расщеплять AP40 на нескольких участках, но основным местом ее действия является связь между Leu(34) и Met(35) (см. рис. 4). ММП-2 и MMP-9 были также обнаружены в астроцитах, окружающих амилоидные отложения в мозге стареющих APP/PS1 и APPsw трансгенных мышей, где они, по-видимому, играют роль в расщеплении Ар (Yan et al., 2006). ММП-9, как и плазмин, способна расщеплять не только мономерные, но и фибриллярные формы Ар. Уровень ММПз и их способность расщеплять Ар изменяется под воздействием различных факторов. Так, в условиях in vivo содержание Ар может быть снижено путем добавления лиганда металлов кликинола и ионов меди (Cu2+), которые повышают уровень экспрессии ММП-2 и ММП-3 (White et al., 2006), что может иметь определенное терапевтическое значение.
Несмотря на наличие убедительной связи между уровнем активности ММПз и амилоидным метаболизмом, в эпидемиологическом исследовании образцов мозга не было выявлено различий в уровне экспрессии и активности ММП-2, -3 и -9 во фронтальной коре при БА и в нормальном мозге (Baig et al., 2007). На генетическом уровне также не было показано наличие корреляций между полиморфизмом генов ММП-3 и ММП-9 и риском развития БА, хотя эти полиморфизмы были связаны с другими заболеваниями (Shibata et al., 2005). С другой стороны, у людей, не являющихся носителями s4 аллели аполипопротеина Е, наблюдается меньший риск развития БА, связанный с определенными полиморфизмами ММР-3 и ММП-9 (Helbecque et al., 2007). Более того, недавно было показано, что защитные эффекты эстрогенов против амилоидной токсичности обусловлены активацией ММП-2 и -9 и соответствующим повышением катаболизма Ар (Merlo, Sortino, 2012).
Катепсин В. За последние годы накоплено довольно большое число данных о роли лизосомальной сериновой протеиназы, кетепсина В, в метаболизме АРР и Ap пептида. Этот фермент был предложен в качестве альтернативной Р-секретазы, действующей преимещественно на нативные (генетически не модифицированные, как в случае семейных форм БА) молекулы АРР (Wang et al., 2012). Также было показано, что ингибиторы катепсина B снижают когнитивный дефицит и понижают содержание Ap в мозге мышей с моделированием БА (Hook et al., 2010). Это согласуется с данными исследований, свидетельствующих, что нейропротекторные свойства катепсина В обусловлены его способностью расщеплять Ap, в частности AP42. Напротив, на модели катепсин В-трансгенных мышей, лишенных этого гена и экспрессирующих APP человека с семейными мутациями, было показано повышение накопления AP42, сенильных бляшек и когнитивный дефицит (Mueller-Steiner et al., 2006). Также недавно было установлено, что катепсин B способен расщеплять C-концевые фрагменты АРР, в том числе и AICD, что указвыает на участие эндосо- мально-лизосомной системы в метаболизме АРР и его фрагментов (Asai et al., 2011).
Действие катепсина В на AP42 сходно с таковым у ACE, поскольку он проявляет активность дикарбоксипептидазы с оброазованием сначала AP40, а потом АР38, тем самым снижая токсичность амилодного пептида (Mueller-Steiner et al., 2006). В результате эндопептидазной активности катепсина В также образуется АР33. Этот фермент также способен расщеплять олигомеры Ap и его фибриллы и действовать как внутри нервных клеток (в лизосомах), так и снаружи, поскольку он интенсивно секретируется активированной микроглией, в частности в ответ на действие Ар (Wu et al., 2013). В связи со сложившимися представлениями о роли катепсина В в расщеплении амилоидного пептида было высказано предположение, что повышения его активности в ткани мозга можно добиться путем инактивации его эндогенных ингибиторов, в частности цистатина С. Действительно, при нокауте гена цистати- на 3 (CST3) у hAPP-J20 трансгенных мышей наблюдалось значительное снижение уровня растворимого Ap, в частности AP42, и сенильных бляшек. Удаление гена цистатина С приводило не только к нормализации Ap-ассоциированного когнитивного дефицита и нарушений в поведении, но также к восстановлению синаптической пластичности в гиппокампе (Sun et al., 2008). К удивлению, оверэкспрессия цистатина C у трансгенных hAPP-J20 мышей также приводила к снижению уровня сенильных бляшек, но, как оказалось, это осуществлялось по механизму, не связанному с активностью катепсина В, а путем взаимодействия цистатина С непосредственно с самим Ap, что препятствовало образованию его токсичных олигомеров и фибрилл (Kaeser et al., 2007; Mi et al., 2007).
Генетические исследования показали наличие связи между полиморфизмом гена цистатина С, который снижает его секрецию, и риском развития спорадической формы БА, что явилось первым свидетельством наличия аутосомно-рецессив- ной аллели, связанной с риском развития этой формы БА (Crawford et al., 2000). Позднее это было подтверждено данными системного метаанализа (Bertram et al., 2007). Исследования катепсина B и цистатина C указывают на важность анализа как каждого индивидуального фермента, так и системы его эндогенных ингибиторов для регуляции метаболизма Ap. Это позволит с большей точностью идентифицировать наиболее перспективные терапевтические мишени для профилактики и лечения БА с минимальными побочными эффектами.
Другие ферменты. Несмотря на довольно широкий спектр амилоид-дегради- рующих ферментов, обнаруженных к настоящему времени, их число продолжает расти. Еще одним физиологически значимым кандидатом на эту роль является пеп- тидасома PreP (Falkevall et al., 2006), которая регулирует уровень содержания Ap в митохондриях. PreP является родственной по свойствам, но не идентичной ИДФ, и принимает участие в расщеплении различных коротких митохондриальных пептидов, в том числе и Ар на нескольких участках его молекулы (см. рис. 4). При БА и у трансгенных мышей в митохондриях клеток мозга наблюдается накопление Ap, что приводит к снижению активности митохондриальных ферментов и угнетению функций митохондрий. Накопление Ap в митохондриях, в основном в его более патогенной форме AP42, наблюдается задолго до образования внеклеточных отложений амилоидных фибрилл и является возможной причиной изменения клеточных функций и их гибели при БА (Caspersen et al., 2005). Хотя до настойщего времени не было выявлено никаких ассоциаций между полиморфизмами гена PreP и риском развития БА, пониженная протеолитическая активность PreP была отмечена в митохондриях мозга при БА по сравнению с контролями того же возраста (Alikhani et al., 2011).
Совершенно неожиданно среди потенциальных кандидатов на роль амилоид- деградирующих ферментов оказался основной белок миелина (MBP) (Liao et al., 2009), который, как полагают, обладает эндогенной активностью сериновых протеаз, приводящей к аутокаталитическому расщеплению его белковой молекулы. Как было показано, MBP также способен связываться с Ap, предотвращая его агрегацию (Hoos et al., 2009).
К числу ферментов, которые могут представлять терапевтический интерес для нейропротекции, также относят цинк-зависимую эндопептидазу, аминопептидазу A (AP-A). Этот фермент специфично удаляет N-концевые аминокислотные остатки и играет важную роль в центральной регкуляции кровяного давления посредством превращения ангиотензина II в ангиотензин III. Поскольку содержание укороченных с N-конца молекул Ap, в частности AP2 существенно повышено при БА и они обладают высокой токсичностью (Wiltfang et al., 2001), снижение активности АР-А может быть терапевтически выгодно. С другой стороны, при действии AP-A образуется субстрат другой аминопептидазы (АР-N), действие которой приводит к доступности остатка N-концевого глутамина для циклизации при участии глутами- нил-циклазы, продукты которой тоже очень токсичны (Schilling et al., 2008). Инактивация AP-A, таким образом, может иметь нейропротекторный характер, препятствуя инициации каскада протеолитических реакций, высвобождающих целый ряд токсичных N-трункированных форм амилоидного пептида.
Еще одна полифункциональная цинк-зависимая эктопептидаза, а именно глу- тамат-карбоксипептидаза II (GCP II), в ткани мозга приводит к расщеплению широко распространенного нейротрансмиттера N-ацетил-аспартил-глутамат (NAAG). Последний, как было показано, вовлечен в патогенез ряда неврологических состояний, и ингибиторы этого фермента имеют нейропротекторные свойства (Neale et al., 2011). В мозге человека этот фермент вырабатывается в основном в астроцитах (Sacha et al., 2007) и, как было показано, способен расщеплять AP40 и AP42 с образованием AP1-14, AP1-1g и AP1-35, которые не обладают токсичностью. Введение специфического ингибитора GCPII трансгенным мышам, моделирующим БА, приводило к повышению уровня Ap. Напротив, повышение экспрессии GCP II в нейронах и глии способствовало снижению образования Ap и его токсичности. Это позволяет рассматривать GCP II как еще одного функционального участника контроля за уровнем накопления Ap в условиях in vivo. Более того, этот фермент способен снижать уровень не только мономерных, но и олигомерных форм Ap и его фибрилл, а также сенильных бляшек (Kim et al., 2010), что позволяет рассматривать его как потенциально важную терапевтическую мишень, хотя более детальные клинические исследования еще ждут своего проведения.
Другие агенты неферментативной природы. Нет никаких сомнений в том, что помимо классических протеолитических ферментов существуют и другие агенты, способные ускорять катаболизм Ap in vivo. Например, было показано, что два фрагмента легких цепей антител также обладают протеолитической активностью по отношению к Ap (Rangan et al., 2003). Первый из них демонстрирует активность, подобную а-секретазе (с23.5), и ведет к образованию 1-16 и 16-40 фрагментов Ар (см. рис. 4). Второй фрагмент обладает карбоксипептидазной активностью, отщепляя последовательно по одной аминокислоте от С-концевой части молекулы Ap. В дополнение к этому, расщепление AP40 фрагментом легкой цепи антител hk14 приводит к изменению его агрегационных свойств и нейтрализует цитотоксический эффект Ap (Liu et al., 2004). Напротив, расщепление AP40 фрагментом легкой цепи антител c23.5 ведет к увеличению скорости агрегации образующихся продуктов, не увеличивая при этом их токсичность в условиях in vivo. Результаты этих исследований свидетельствуют, что фрагменты антител способствуют протеолитической деградации Ap и, таким образом, имеют потенциальную терапевтическую значимость для контроля уровня образования, агрегации и токсичности Ap в условиях in vivo. С другой стороны, эти данные заставляют с осторожностью относиться к предлагаемому методу контролируемого образования Ap путем повышения активности а-секретазы, поскольку это может вести к увеличению агрегационных свойств амилоидного пептида с образованием более токсичных олигомеров.
Еще одним фактором, способствующим снижению уровня эндогенного Ap, является глюкагонподобный пептид (GLP-1) (Perry, Greig, 2004). Локализация рецептора GLP в головном мозге человека и грызунов коррелирует с центральной ролью этого пептида в регуляции потребления пищи, однако также было показано, что стимуляция нейрональных GLP-1-рецепторов регулирует нейрональную пластичность и выживаемость нейронов. GLP-1 является естественным аналогом экзендина-4, который, как было установлено, снижает уровень эндогенного Ap в паренхиме мозга мышей и уровень APP в нейронах. Поскольку GLP-1 активирует рецепторы, сопряженные с Gs белками, его действие посредством индукции синтеза цAMФ, может влиять на многие функции клетки, в том числе на их выживаемость. Это делает GLP-1 и другие агонисты рецепторов 2 класса, сопряженных с
G-белками, потенциально важными агентами для развития новых стратегий лечения БА (Martin et al., 2005).
Помимо ферментов в поддержании амилоидного гомеостаза в ткани мозга принимают также участие белки-переносчики, которые способны связывать и выводить Ap по периваскулярным путям. К их числу относятся аполипопротеины (АроЕ), связь которых с патогенезом БА уже рассматривалась выше, но точный механизм действия которых пока не установлен (для обзора см. Zlokovic, 2013), а также переносчик тироксина - транстиретин (TTR) (Du et al., 2012). Последний представляет особый интерес, поскольку его регуляция в нервных клетках осуществляется сходным с неприлизином образом (Kerridge et al., submitted). TTR, в основном образуется в сосудистом сплетении мозга (Sousa et al., 2007), но он также может экспрессироваться и нервными клетками (Li et al., 2010). В литературе есть указание на то, что TTR может проявлять пептидазную активность, но функциональный вклад этого белка в качестве фермента в катаболизм Ap еще нуждается в подтверждении (Costa et al., 2008). Tем не менее, имеются убедительные данные о роли TTR в защите нервных клеток от повреждающего действия Ap и восстанавле- нии когнтитивных свойств у животных (Buxbaum et al., 2008; Brouillett et al., 2012). С возрастом и при БА уровень TTP в ткани мозга существенно снижается (Tsai et al., 2009), что указывает на актуальность изучения механизмов регуляции его экспрессии, а также поиск соединений, способствующих нормализации его функций.
2.6.
Еще по теме Амилоид-деградирующие ферменты:
- Регуляция свойств амилоид-деградирующих ферментов
- Характеристика сывороточного амилоида А
- Локализация амилоида. Вопросы патогенеза.
- Роль в-амилоида в патогенезе БА
- в-амилоид и периферические нарушения: вклад в нейродегенеративный процесс
- Роль амилоида-р в болезни Альцгеймера
- Измерение концентрации амилоида А и CA125 методом иммуноферментного анализа
- Белки предшественники амилоидов
- Ферменты процессинга Ар
- Ферменты пищеварительного тракта
- Ферменты пищеварительного тракта
- 9.0.5. Биологическая регуляция ферментов