Патогенез.
Существующие теории патогенеза атеросклероза можно свести к двум, принципиально отличающимся по своим ответам на вопрос: что первично, а что вторично при атеросклерозе, другими словами, что является причиной, а что следствием - липоидоз внутренней оболочки артерий или дегенеративно-пролиферативные изменения последней.
Этот вопрос впервые был поставлен P Вирховым (1856). Он же первый и ответил на него, указав, что "при всех условиях процесс, вероятно, начинается с определенного разрыхления соединительнотканного основного вещества, из которого большей частью состоит внутренний слой артерий".
C тех пор и берет начало представление немецкой школы патологов и ее последователей в других странах, согласно которому при атеросклерозе первоначально развиваются дистрофические изменения внутренней оболочки стенки артерий, а отложение липидов и солей кальция - явление вторичного порядка. Преимуществом данной концепции является то, что она в состоянии объяснить развитие спонтанного и экспериментального атеросклероза как в тех случаях, когда имеются выраженные нарушения холестеринового обмена, так и при их отсутствии. Первостепенную роль авторы указанной концепции отводят артериальной стенке, т. е. субстрату, который непосредственно вовлекается в патологический процесс. "Атеросклероз является не только и даже не столько отражением общих обменных сдвигов (лабораторно они могут быть даже неуловимы), сколько производным собственных структурных, физических и химических превращений субстрата артериальной стенки... Первичный фактор, ведущий к атеросклерозу, лежит именно в самой артериальной стенке, в ее структуре и в ее энзимной системе" [Давыдовский И. В., 1966].
В противоположность этим взглядам со времени опытов Н. Н. Аничкова и С. С. Халатова, главным образом благодаря исследованиям отечественных и американских авторов, успешно развивается концепция о роли в развитии атеросклероза общих метаболических нарушений в организме, сопровождающихся гиперхолестеринемией, гипер- и дислипопротеидемией.
C этих позиций, атеросклероз - следствие первичной диффузной инфильтрации липидов, в частности холестерина, в неизмененную внутреннюю оболочку артерий. Дальнейшие изменения в сосудистой стенке (явления мукоидного отека, дистрофические изменения волокнистых структур и клеточных элементовподэндотелиального слоя, продуктивные изменения) развиваются в связи с наличием в ней липидов, т. е. являются вторичными.
Первоначально ведущая роль в повышении уровня липидов, особенно холестерина, в крови приписывалась алиментарному фактору (избыточному питанию), что дало название и соответствующей теории возникновения атеросклероза - алиментарной. Однако очень скоро ее пришлось дополнить, так как стало очевидным, что не все случаи атеросклероза можно поставить в причинную связь с алиментарной гиперхолестеринемией.
Согласнокомбинационной теории Н. Н. Аничкова, в развитии атеросклероза, кроме алиментарного фактора, имеют значение эндогенные нарушения липидного обмена и его регуляции, механическое влияние на стенку сосуда, изменения артериального давления, главным образом его повышение, а также дистрофические изменения в самой артериальной стенке. В этой комбинации причин и механизмов атерогенеза одни (алиментарная и/или эндогенная гиперхолестеринемия) играют роль инициального фактора. Другие либо обеспечивают увеличенное поступление холестерина в стенку сосуда, либо уменьшают его экскрецию из нее через лимфатические сосуды.
В крови холестерин содержится в составе хил омикронов (мелкодисперсных частиц, не растворенных в плазме) и липопротеидов - надмолекулярных гетерогенных комплексов триглицеридов, эфиров холестерина (ядро), фосфолипидов, холестерина и специфических белков (апопротеиды: АПО А, В, С, Е), образующих поверхностный слой. Существуют определенные отличия липопротеидов по размерам, соотношению ядра и оболочки,
качественному составу и атерогенности.
Выделены 4 основные фракции липопротеидов плазмы крови в зависимости от плотности и электрофоретической подвижности.
Обращает на себя внимание высокое содержание белка и низкое - липидов во фракции липопротеидов высокой плотности (ЛПВП - ?-липопротеиды) и, наоборот, низкое содержание белка и высокое - липидов во фракциях хиломикронов, липопротеидов очень низкой плотности (ЛПОНП - пре- ? -липопротеиды) и липопротеидов низкой плотности (ЛИНИ - ?-липопротеиды).
Таким образом, липопротеиды плазмы крови осуществляют доставку синтезированных и полученных с пищей холестерина и триглицеридов к местам их использования и депонирования.
ЛПВП оказывают антиатерогенное действие путем обратного транспорта холестерина из клеток, в том числе из сосудов, к печени с последующим выведением из организма в форме желчных кислот. Остальные фракции липопротеидов (особенно ЛИНИ) являются атерогенными, обусловливая избыточное накопление холестерина в стенке сосудов.
В табл. 5 приведена классификация первичных (генетически обусловленных) и вторичных (приобретенных) гиперлипопротеидемий с той или иной степенью выраженности атерогеннош действия. Как следует из таблицы, в развитии атероматозных изменений сосудов основную роль играют ЛИНИ и ЛПОНП, их повышенная концентрация в крови, избыточное поступление в интиму сосудов.
Избыточный транспорт ЛИНИ и ЛПОНП в сосудистую стенку прошествует повреждению эндотелия.
В соответствии с концепцией американских исследователей И Голдстайна и М. Брауна, Л ПИП и ЛПОНП в клетки поступают путем взаимодействия со специфическими рецепторами (АПО В, Е-реиепторы-гликопротеиды), после чего происходит их эндоцитозный захват и слияние с лизосомами. При этом Л ПИП расщепляются на белки и эфиры холестерина. Белки расщепляются на свободные аминокислоты, которые покидают клетку. Эфиры холестерина подвергаются гидролизу с образованием свободного холестерина, который поступает из лизосом в цитоплазму с последующим использованием для тех или иных целей (образование мембран, синтез стероидных гормонов и т. д.). Важно, что этот холестерин угнетает его синтез из эндогенных источников, при избытке образует "запасы" в форме эфиров холестерина и жирных кислот, но, самое главное, по механизму обратной связи угнетает синтез новых рецепторов для атерогенных липопротеидов и их дальнейшее поступление в клетку.
Наряду с регулируемым рецепторопосредованным механизмом транспорта ЛП, обеспечивающим внутренние потребности клеток в холестерине, описан межэндотелиальный транспорт, а также так называемый нерегулируемый эндоцитоз, который представляет собой трансцеллюлярный, в том числе трансэндотелиальный везикулярный транспорт Л11Ш1 и ЛПОНП с последующим экзоцитозом (в интиму артерий из эндотелия, макрофагов, гладкомышечных клеток).C учетом изложенных представлений механизм начального этапа атеросклероза, характеризующегося избыточным накоплением липидов в интиме артерий, может быть обусловлен:
1. Генетической аномалией рецептор-опосредованного эндоцитоза Л11Ш1 (отсутствие рецепторов - менее 2% от нормы, уменьшение их числа - 2 - 30% от нормы). Наличие таких дефектов обнаружено при семейной гиперхолестеринемии (гипербеталипопротеидемия II А типа) у гомо- и гетерозигот Выведена линия кроликов (Ватанабе) с наследственным дефектом рецепторов к JIIIHII.
2. Перегрузкой рецепторопосредованного эндоцитоза при алиментарной гиперхолестеринемии. И в том, и в другом случае наступает резкое усиление нерегулируемого эндоцитознош захвата частиц ЛП эндотелиальными клетками, макрофагами и гладкомышечными клетками стенки сосудов вследствие выраженной гиперхоле стеринемии.
3. Замедлением удаления атерогенных липопротеидов из стенки сосудов через лимфатическую систему в связи с гиперплазией, гипертензией, воспалительными изменениями.
Существенный дополнительный момент - различные превращения (модификации) липопротеидов в крови и сосудистой стенке. Речь идет об образовании в условиях гиперхолестеринемии аутоиммунных комплексов ЛП - IgG в крови, растворимых и нерастворимых комплексов ЛП с гликозаминогликанами, фибронектином, коллагеном и эластином в сосудистой стенке (А. Н. Климов, В. А. Нашрнев).
По сравнению с нативными ЛП захват модифицированных ЛП клетками интимы, в первую очередь макрофагами (с помощью нерегулируемых холестерином рецепторов), резко возрастает. Это, как полагают, является причиной превращения макрофагов в так называемые пенистые клетки, которые составляют морфологическую основу стадии липидных пятен и при дальнейшем прогрессировании -атером.
Миграция кровяных макрофагов в интиму обеспечивается с помощью моноритарного хемотаксическош фактора, образующегося под действием ЛП и интерлейкина-1, который выделяется из самих моноцитов.На заключительном этапе формируются фиброзные бляшки как ответ гладкомышечных клеток, фибробластов и макрофагов на повреждение, стимулируемый факторами роста тромбоцитов, эндотелиоцитов и гладкомышечных клеток, а также стадия осложненных поражений - кальцификация,тромбообразование и др. (рис. 19.13).
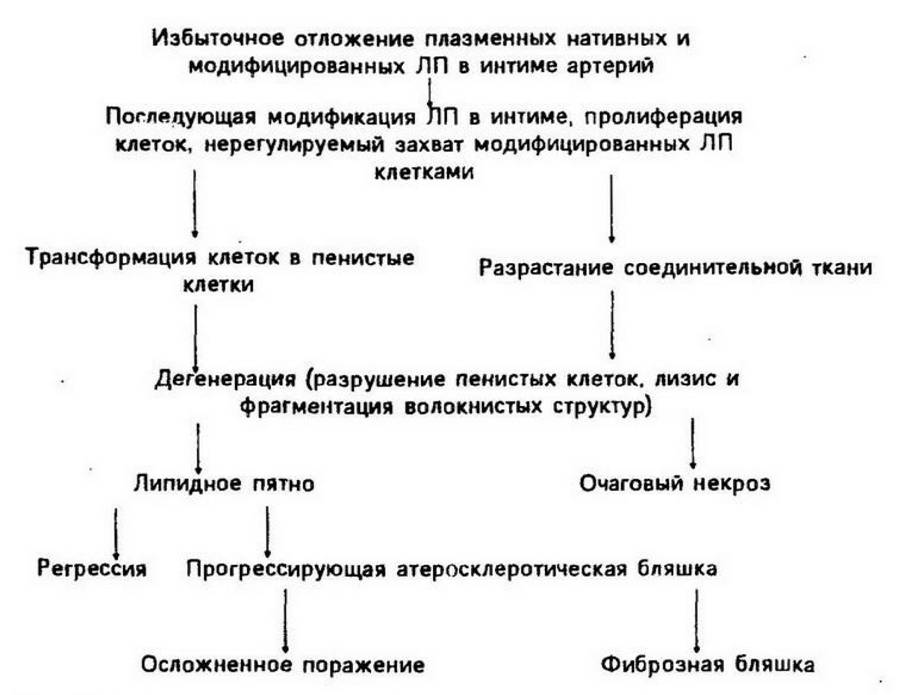
Рис. 19.13. Патогенез атеросклероза (по В, Л. Нагорневу, А. Д, Денисенко, 1989).
Приведенные выше концепции патогенеза атеросклероза имеют свои сильные и слабые стороны. Наиболее ценным достоинством концепции общих метаболических нарушений в организме и первичного липоидоза артериальной стенки является наличие экспериментальной холестериновой модели. Концепция первичного значения местных изменений в артериальной стенке, несмотря на то что была высказана более 100 лет назад, пока не имеет убедительной экспериментальной модели.
Как видно из изложенного, в целом они могут дополнять друг друга.
Еще по теме Патогенез.:
- Учение о патогенезе
- Патогенез
- Патогенез.
- 3. ПАТОГЕНЕЗ
- Патогенез тромбозов при АФС
- Патогенез.
- ЭТИОЛОГИЯ И ПАТОГЕНЕЗ
- Патогенез
- Основные гипотезы патогенеза БА
- Общие вопросы учения о патогенезе болезней
- Патогенез
- Патогенез
- Патогенез.
- Патогенез.