2.5. Пероксигеназные процессы и лейкозогенез
Развитие лейкозов при лучевых и других лейкозогенных воздействиях, как известно, связано со злокачественным системным заболеванием кроветворной ткани, нарушением кроветворения, выражающемся в разрастании незрелых патологических клеточных элементов.
В зависимости от того, какие из этих элементов преобладают, различают разные виды этой патологии. Общепризнанным считается представление, согласно которому наиболее реальными клетками-мишенями для лейкозной трансформации являются полипотентные стволовые кроветворные клетки (ПСКК) и клетки-предшественники отдельных ростков кроветворения. Однако причины высокой чувствительности данной категории клеток в различным лейкозогенным факторам достоверно всё ещё не установлены, и, во всяком случае, единого мнения на этот счёт у исследователей пока нет.По развиваемой нами концепции, лейкозогенез как один из видов канцерогенеза возникает при наличии в клетке дисбаланса ∆Л (ПО – АО), аналогичного по сути (по значениям и вызываемым им последствиям) ∆К (ПО – АО) для негемопоэтических клеток опухоли. Почему и как создаётся такое состояние в гемопоэтических клетках это – принципиальные вопросы, на которые желатель-но найти ответы, чтобы обосновать кислородно-перекисный механизм лейкозогенеза. В данном параграфе мы попытались высказать по этой теме некоторые соображения, представляющиеся достаточно логичными и разумными.
2.5.1. С точки зрения кислородно-перекисной концепции, для ПСКК и ство-ловых клеток в начальных стадиях коммитирования в определённом направ-лении дифференцировки должны быть характерны относительно гипероксические условия. Последние могут быть созданы, прежде всего, за счёт малого числа действующих митохондрий. Такие принципиально важные данные в обо-бщённом виде нам пока не встретились, но отдельные факты здесь известны. На низкое содержание митохондрий в стволовых и эмбриональных клетках указывается, например, в работе, авторы которой (Von Wagenheim, Peterson, 1998), опираясь на этот факт, рассматривают механизм контроля клеточной пролиферации с помощью процесса дифференцировки в связи с возможными механизмами старения, канцерогенеза и апоптоза.
Сходная кислородно-перекисная ситуация может возникнуть и в случае сосредоточения митохондрий (при достаточном даже их количестве) преимущественно в околоядерной зоне, где эти органеллы относительно примитивны в структурно-функциональном отношении, не являются полноценными «устройствами» для генерации ATP и активного потребления О2 (Озернюк, 1978; Ступина и др., 1993; см. также п. 3.6.2).Прежде чем продолжить лейкозную тему, представляется важным остановиться на вопросе о состоянии и специфике энергетического обмена в зрелых форменных элементах крови, в частности, в моно- и полинуклеарных фагоцитах, предполагая, что для их недифференцированных предшественников характерна та же энергетика. Функции фагоцитов и такой их феномен как «окислительный (дыхательный) взрыв» определяются структурно-функциональными и некоторыми другими их особенностями. Так, у данных видов лейкоцитов отсут-ствуют митохондрии или их очень мало (Мусил, 1985; Маянский и др., 1999), а они, как известно, являются основными потребителями О2 в клетке. В этой ситуации почти автоматически должны обеспечиваться низкий уровень утилизации О2 и соответственно относительная внутрифагоцитарная гипероксия, пос-кольку скорость потребления О2 пропорциональна количеству митохондрий в клетке (Ames et al., 1995).
Подобная информация приводилась в литературе и раньше. Например, отмечалось, что по сравнению с большинством других клеток животного организма вообще всем лейкоцитам присущи не интенсивное дыхание, а интенсивный гликолиз. Об этом свидетельствуют величины отношений коэффициентов Варбурга QCO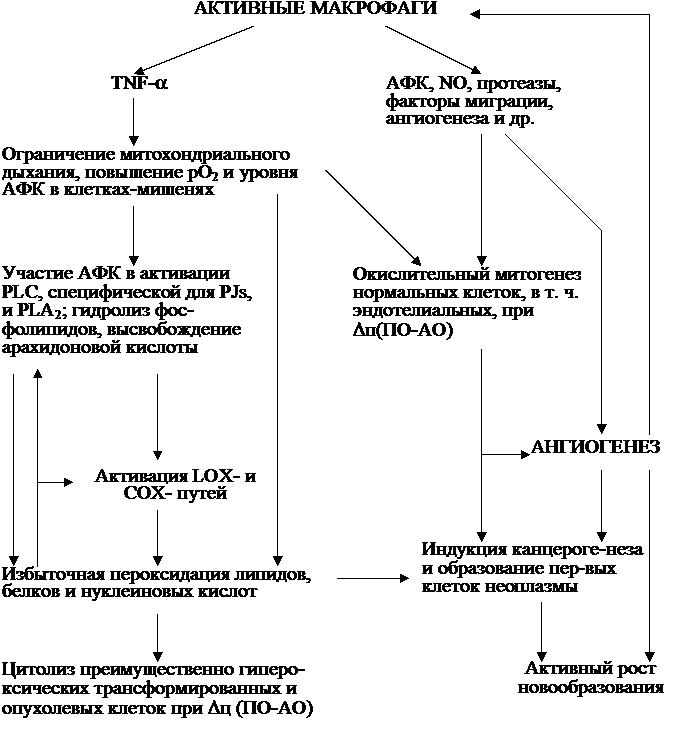 и QO: для большинства клеток взрослой ткани их отноше-ние < 1, для лейкоцитов же, как и для клеток эмбриональной и раковой тканей, эта величина всегда > 1, что согласуется со сравнительно малым числом мито-хондрий в белых клетках крови, особенно в лимфоцитах (Луганова, Сейц, 1958; Леонтович, 1976).
и QO: для большинства клеток взрослой ткани их отноше-ние < 1, для лейкоцитов же, как и для клеток эмбриональной и раковой тканей, эта величина всегда > 1, что согласуется со сравнительно малым числом мито-хондрий в белых клетках крови, особенно в лимфоцитах (Луганова, Сейц, 1958; Леонтович, 1976).
Указанные приспособительные изменения имели определённый биологи-ческий смысл, и сейчас кажется очевидным, что они понадобились в ходе эволюции для выполнения фагоцитами защитных функций путём выработки концентрированного «окислительного удара» против инородных тел в орга-низме.
Стимуляция фагоцитов вызывает всплеск независимого от митохон-дриального дыхания потребления О2, приводящий к образованию и секреции повышенного количества высокоактивных прооксидантов О , Н2О2, ОН˙.
, Н2О2, ОН˙.По современным представлениям, функцию синтеза АФК выполняет активируемая PKC и арахидоновой кислотой NADPH-оксидаза – специализированный ферментный комплекс, локализованный, по некоторым данным, на внутренней поверхности плазматической мембраны (Дубинина, 1989; Shiose, Sumi-moto, 2000). NADPH-оксидаза катализирует реакцию окисления NADPH до NADP+ и одновременно одноэлектронного восстановления О2 до О путём переноса электронов от NADPH с внутренней стороны мембраны к О2 на наружной её стороне (Клюбин, Гамалей, 1997; Babior, 1999):
2 NADPH + 4 O2 → 2 NADP+ + 4 О + 2 H+.
В данном варианте механизма речь идёт об использовании внеклеточных молекул О2 для синтеза АФК на внешней поверхности плазматической мемб-раны фагоцитов, т. е. фактически вне этих клеток. Однако образование повышенного количества АФК будет, очевидно, недостаточно эффективным, если приток О2 к клеткам по той или иной причине станет неустойчивым и ненадёжным. Мы полагаем поэтому, что должен существовать дополнительный механизм, поддерживающий образование и секрецию АФК за счёт создания необходимых для этого условий внутри самих фагоцитов. Одним из основных таких условий должна быть указанная выше внутрифагоцитарная гипероксия, которая, естественно, невозможна в присутствии множества конкурирующих за кис-лород митохондрий. Генерируемые О и его производные, в частности Н2О2, могут свободно проникать из клетки через мембрану во внеклеточное пространство и в кровяное русло (Morre et al., 2000), причём скорость их диффузии через плазматическую мембрану зависит от типа клеток и находится в пределах 0,02-0,4 см/мин (Сидорик и др., 1989).
Ввиду ограниченности митохондриального дыхания все необходимые энергетические затраты в фагоцитах обеспечиваются в основном за счёт гликолиза. Так, трансформация моноцитов в макрофаги сопряжена с возрастанием утилизации глюкозы и продукции молочной кислоты. Даже в аэробных условиях 90 % энергии для фагоцитоза полиморфноядерные лейкоциты получают не от дыхания, а в результате гликолиза, поэтому ингибиторы гликолиза (йодацетат, фторид) легко и полностью блокируют в них фагоцитоз и другие клеточные процессы. Ими блокируется также цитолиз клеток-мишеней лимфоцитами-киллерами (Мохова, 1987). Кстати, глюкоза необходима и для функционирования NADPH-оксидазы: при активации последней падает концентрация NADPH в клетке, что стимулирует окисление глюкозы через гексозо-монофосфатный шунт, поставляющий NADPH в фагоцитах (см. Миллер и др., 2002).
Уменьшение общей мощности митохондрий в микро- и макрофагах и, веро-ятно, в их предшественниках, создавая в принципе условия для накопления О2 и использования его в целях поддержки «кислородного взрыва» изнутри фагоцитов, одновременно придаёт указанным клеткам такие качества, как повышенная радиочувствительность, приближенность к состоянию развития избыточного ПОЛ. Следовательно, в данном случае новая полезная для организма функция приобретена моно- и полинуклеарными фагоцитами в какой-то мере в ущерб их собственной неприкосновенности – ведь с точки зрения кислородно-пере-кисной концепции онкогенеза они уже «подготовлены» к восприятию канцерогенных воздействий и злокачественной трансформации, если находятся в сос-тоянии активной пролиферации.
Изложенным представлениям, казалось бы, противоречат сведения о том, что альвеолярные макрофаги в отличие от других их типов имеют хорошо развитый аппарат митохондрий (Маянский, 1991). Но это исключение естественно объяснить опять-таки адаптацией лёгочных макрофагов к функционированию их при высоком рО2: утилизация О2 увеличенным количеством митохондрий несколько снижает слишком опасный уровень внутриклеточной гипероксии и тем самым защищает макрофаги от их неминуемого окислительного разрушения.
Поскольку по своему смыслу данная адаптация противоположна другой – созданию достаточной внутрифагоцитарной гипероксии для реализации «кис-лородного взрыва», то при установлении количества и размеров митохондрий достигается, по-видимому, определённый компромисс, позволяющий достичь обе указанные цели.Представленная выше ситуация в моно- и полинуклеарных фагоцитах пред-намеренно рассматривается нами в упрощенном варианте, чтобы оттенить в них постулируемые принципиальные структурно-функциональные перестройки и связанные с ней последствия. В действительности же взаимосвязь многих внут-риклеточных процессов, естественно, намного сложнее. Так, в неактивированном макрофаге большинство поглощённых им молекул О2, по-видимому, связывается специальными депонирующими соединениями (предположительно, каротиноидами и ретиноидами), и в этом состоянии эффекторная клетка должна быть радиорезистентной. Однако после активации в момент быстрого высвобождения депонированного О2, используемого для образования АФК и оксиповреждения чужеродных веществ, макрофаг становится гипероксичным и, следовательно, радиочувствительным.
Неизвестно, начинаются ли указанные выше перестроения уже на уровне предшественников дифференцированных лейкоцитов и, в частности, на стадии бластных клеток или же они присущи им изначально. Не исключено, что приспособительный характер утраты ими всех или части митохондрий для выполнения новых функций, в данном случае эффекторных, в принципе сходен с адаптивным процессом самоликвидации митохондрий при созревании эритробластов в эритроциты с целью свободной, бесконкурентной реализации в них «гемоглобинового» механизма депонирования и транспорта О2. Однако высокая радиочувствительность стволовых клеток костного мозга и пролиферирующих бластных клеток как будто бы указывает на то, что некоторые структурно-функциональные особенности, придающие им это О2-зависимое свойство, уже присутствуют в них.
По названным, вероятно, причинам ПСКК присуща хорошо известная исследователям повышенная чувствительность к различным лейкозогенным воздействиям (Бутенко и др., 1984) и, в частности, высокая радиочувствите-льность (Чертков, Гуревич, 1984; Москалев, 1991).
Косвенным признаком повышенного рО2 и ПОЛ в нормальной кроветворной клетке костного мозга, обладающей практически неограниченной способностью к пролиферации, слу-жат данные об очень высоком содержание в ней cGMP (Федоров и др., 1990). Действительно, оксиданты различной природы (О2 воздуха, радикал ОН˙, гидроперекиси липидов, продукты метаболизма арахидоновой кислоты и др.) активируют гуанилатциклазу и, следовательно, повышают концентрацию cGMP, причастного к пролиферации клеток вообще и ПСКК в частности (см. п. 2.3.2).При лейкозной трансформации митохондрии в ПСКК и костномозговых клетках-предшественниках гемо- и лимфопоэза, как правило, подвергаются зна-чительным негативным изменениям, что, в частности, выражается в набухании митохондриального матрикса, дезорганизации крист, разрыве оболочки митохондрий и пр. (Schumacher et al., 1980). По данному признаку лейкозные клетки не отличаются от неопластических другого вида. Гетерогенность популяции ПСКК и, следовательно, различие в них постулируемых пероксигеназных условий не могут привести к одновременному тотальному поражению всей популяции. Каждая из её клеток будет изменяться по «индивидуальному плану», а какая-то часть популяции вообще остаётся неповреждённой. При этом пролиферация трансформированных клеток становится относительно независимой от их микроокружения.
Лейкозогенез, таким образом, предлагается рассматривать как частный случай общего кислородно-перекисного механизма канцерогенеза (Лю, Шайхутдинов, 1991; Лю, 2002а). И при этой форме патогенеза основным первичным объектом воздействия лейкозогенных факторов представляются митохондрии. Постулируемая нами количественная и/или функциональная недостаточность этих органелл в ПСКК и их потомках предопределяет высокую чувствительность названных клеток к негативным сдвигам в дыхательном аппарате, с которыми связывается увеличение в них дисбаланса ∆ (ПО – АО) с просто «пролиферативного» уровня ∆П (ПО – АО) до «лейкозогенного» ∆Л (ПО – АО).
С позиций теории устойчивости кибернетических систем указанные выше особенности биоэнергетики ПСКК и клеток на ранних стадиях гемопоэза должны означать, что они находятся как бы в состоянии неустойчивого динамического равновесия. Поэтому даже при малых возмущающих воздействиях определённые клетки из гетерогенного их состава могут сравнительно легко сходить с пути нормального кроветворения на развитие апоптоза, лейкозогенеза и других заболеваний их по кислородно-перекисному механизму. Как представляется нам, именно по этой, в основном, причине увеличивается частота возникновения спонтанных лейкозов и ускоряется их развитие у мышей линии AKR под влиянием облучения 137Cs в малой (1,2 – 2,4 сГр) низкоинтенсивной дозе (Бурлакова, Ерохин, 2001).
С учётом сказанного выше об особенностях биоэнергетики в ПСКК, клетках-предшественниках отдельных ростков кроветворения и в фагоцитах вполне реальным представляется и путь индукции лейкозогенеза, связанный с воздействием на кроветворные клетки их микроокружения. Речь идёт о ретикулярных клетках, образующих сетчатую соединительную ткань, которая составляет основу кроветворных органов. Эти клетки относятся к ретикуло-эндотелиаль-ной системе и обладают высокой фагоцитарной способностью. При наличии раздражающих факторов ретикулярные клетки могут превращаться в активные макрофаги. В этом качестве они, продуцируя АФК, способны, очевидно, сравнительно легко увеличить в прилежащих к ним относительно гипероксичных уже кроветворных клетках значение дисбаланса ∆П (ПО – АО) до «лейкозогенного» ∆Л (ПО – АО). Другими словами, здесь достаточно вероятна реализация кислородно-перекисного механизма лейкозогенеза, индуцированного макрофагами самого кроветворного органа. Такое представление не является чем-то удивительным в свете того, что нами в своё время (Лю, Шайхутдинов, 1983, 1986, 1989, 1991) был обоснован «макрофаговый» механизм канцерогенеза, индуцируемого инородными телами (см. также главу 5).
В качестве активаторов клеток ретикулярной соединительной ткани стромы выступают, вероятно, различные колониестимулирующие факторы (CSFs), в частности гранулоцитов (CSF-G) и гранулоцитов-макрофагов (CSF-GM). Влияя на О2-активирующие системы плазматической мембраны этих клеток, CSFs могут усиливать генерацию ими АФК. Во всяком случае, так происходит in vitro при воздействии CSF-G на нейтрофилы, на поверхности которых имеются рецепторы к CSF-G. Показано и другое: «восстановление функциональной активности нейтрофилов у детей с онкогематологическими заболеваниями в процессе терапии с использованием CSF-G происходит не в результате прямого воздействия на нейтрофилы, а на уровне вмешательства в процессы кроветворения в костном мозге» (Коваленко и др., 1998). По данным работы (Pyatt et al., 1996), реактивные производные О2, генерируемые ксантином и ксантинокси-дазой, опосредуют синергизм CSF-GM и фактора стволовых клеток, который усиливает митогенный эффект многих кроветворных факторов роста, в отношении ранних кроветворных родоначальных клеток мыши. Предварительная обработка этих клеток АФК дозозависимо усиливала образование колоний под влиянием CSF-GM, но не CSF-G.
Как представляется нам, прямое или косвенное воздействие CSFs на ПСКК и клетки-предшественники отдельных ростков кроветворения в норме приз-вано стимулировать их окислительный митогенез. Однако в действительности иногда проявляются негативные процессы, вызванные главным образом избыточной продукцией АФК при указанной стимуляции. Одним из таких следствием и может быть излагаемый механизм лейкозогенеза. Известны также и другие повреждающие возможности CSFs. Например, экспрессия гена CSF-GM в клетках рака толстой кишки мыши индуцирует противоопухолевый эффект, что обнаруживается по скоплению нейтрофилов и некротическим изменениям раковой ткани, снижению её онкогенности и увеличению выживаемости животных (Gunji et al., 1996). Здесь, очевидно, проявляется действие одновременно двух индуцируемых CSF-GM факторов: увеличение числа и активности АФК-генерирующих клеток, вследствие чего дисбаланс ∆ (ПО – АО) в опухолевых клетках возрастает до апоптозного ∆А2 или сразу до цитолизного ∆Ц (см. п. 7.1.1).
Следует отметить, что гипотеза о причастности митохондрий в лейкозогенезу выдвигалась и раньше (Szekely et al., 1976), однако, в кислородно-перекисном аспекте роль этих органелл в механизме данной патологии обсуждается, по-видимому, нами впервые. Приводимые ниже материалы призваны дополнить эту концепцию, повысив в её пользу уровень аргументации.
Еще по теме 2.5. Пероксигеназные процессы и лейкозогенез:
- взаимосвязь процессов, ведущих к лейкозогенезу, с дифференцировкой находящихся на ранней стадии развития кроветворных клеток.
- 2.5.2. Повышенный уровень ПОЛ при лейкозогенезе
- 2.3.6. Пероксигеназные условия в опухолевых клетках
- Справедливость процесса или справедливость результата процесса
- Назаренко Г. И., Осипов Г. С.. Основы теории медицинских технологических процессов. Ч. 2. Исследование медицинских технологических процессов на основе интеллектуального анализа данных. - М.: ФИЗМАТЛИТ,2006. - 144 с., 2006
- 1.2.1. Роль ПОЛ в процессах старения
- Частный технологический процесс
- Токсический процесс
- Оценка возможности процесса
- Психические процессы личности
- Патологические процессы и состояния