Наследственные факторы.
Наследственные факторы играют несомненную роль в этиопатогенезе отслойки ретины. Целый ряд наследственных гиалоидоретинопатии связан с ретиношизисом.
Ювенильный ретиношизис, связанный с полом, встречается только у мужчин и характеризуется расслоением сетчатки в нижне-наружных квадрантах с образованием больших овальных или круглых отверстий на внутренней поверхности ретиношизиса.
Характерны дегенерация макулы, частичная атрофия зрительного нерва, относительная центральная скотома, гиперметропия, астигматизм. Этот вид гиалоидоретинопатии наследуется по рецессивному типу.Довольно редко встречается ювенильный ретиношизис аутосомального типа, сочетающийся с расслоением височных половин сетчатки с отложением пигмента. В отличие от ювенильного ретиношизиса, сцепленного с полом при аутосомальном ретиношизисе, макулярная область, как правило, не подвергается дегенерации.
Оба описанных выше типа ретиношизиса следует дифференцировать с гигантскими кистами в посторальной зоне в нижненаружных квадрантах, встречающихся у лиц обоих полов. В настоящее время накоплен целый ряд клинических факторов о семейных характерах пресенильных и сенильных случаях ретиношизиса. Двусторонний характер и зеркальная симметрия ретиношизиса у членов одной и той же семьи явно говорят в пользу генетических факторов, контролирующих локализацию, протяженность, время наступления и прогрессирование этого поражения сетчатки (59,72,76,95,96,173).
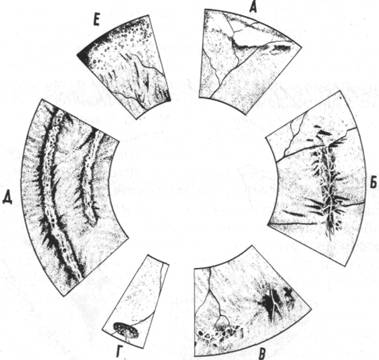
Рис.7а. Схематическое изображение ретинальных дегенераций, являющихся фактором риска в возникновении отслойки сетчатки.
А—дегенерация типа «белого без вдавления»; Б—решетчатая дегенерация; В—пигментная решетчатая дегенерация; Г—ретинальная эрозия; Д—дегенерация типа «следа улитки»; Е—«инееподобная» дегенерация (по Кинелл)
Самую большую группу наследственных изменений сетчатки составляют аутосомальные регматогенные хориоретиногиалоидопатии (рис.7а).
К ним относится, прежде всего, так называемая дегенерация типа «следа улитки» (59,86,136). Этот вид дегенерации описывается также под названием простой опоясывающей атрофии сетчатки (130,141). Наиболее важной с клинической точки зрения является т.н. решетчатая дегенерация (99,199,204,206). Дегенерация имеет четкую клиническую картину в виде белых линий, в совокупности напоминающих собой веточку, и которая является обычно частью запустевших и облитерированных веточек ретинальных сосудов. В местах этой дегенерации гистологически, как правило, выявляется прочное витреоретинальное сращение. Одна из самых ранних описанных дегенерации носит название Фогта-Блессига-Иванова и представляет собой кистовидную дегенерацию в виде микрокист в атрофических зонах сетчатки, как правило, на крайней периферии. Явно наследственный характер носит также такой вид дегенерации, как филяриформные или нитевидные дегенерации, описанные Томсоном и соавт. (цит.106). Довольно часто встречается такой доброкачественный тип дегенерации, как постэкваториальная хориоретинальная атрофия, часто сочетающаяся с другими видами гиалоидоретинопатии, в частности типа Вагнера, и сопровождающаяся пигментацией по ходу ретинальных сосудов на крайней периферии (рис.7б). Наследственный характер носят также абиотрофические хориоретиногиалоидопатии на глазах с высокой близорукостью, проявляя зачастую аутосомальный доминантный тип наследования отслоек сетчатки. Частота отслоек сетчатки на глазах с высокой близорукостью составляет от 4 до 6,8% (61,77,80,121,137,142). Сравнивая частоту отслойки сетчатки 0,01% в нормальной популяции до возраста 80 лет, можно заметить четкую статистическую разницу между двумя цифрами.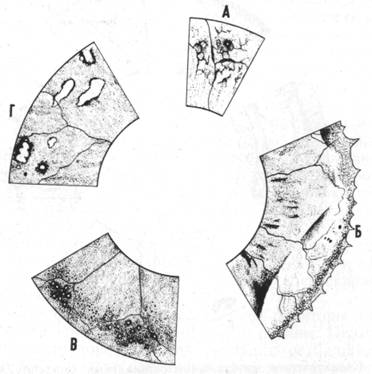
Рис.7б. Схематическое изображение относительно доброкачественных ретинальных дегенераций.
А — дегенерация Вагнера — не специфическая ретикулярная пигментная дегенерация; Б — кистевидная дегенерация и ретиношизис; В — дегенерация сетчатки в виде друз; Г — дегенерация сетчатки типа «булыжной мостовой» (по Кинелл)
Целый ряд наследственных гиалоидоретинальных дегенерации носит имя авторов, впервые их описавших.
Такова, например, гиалоидоретинальная дегенерация типа Вагнера, характеризующаяся прогрессирующим пресенильным образованием катаракты, периферической пигментной ретинальной дегенерацией, выраженными витреальными изменениями в виде разжижения стекловидного тела, образованием лентовидных мембран, снижением зрения к концу четвертого десятилетия жизни из-за катаракты и ретинальных изменений, сужением поля зрения, снижением электроретинограммы и нормальным сумеречным зрением. Синдром Вагнера следует различать от другого, близкого по происхождению синдрома Гольдмана-Фавра, описанного также под названием идиопатического ретиношизиса с ранней гемералопией (63,96,109). К характерным чертам его относятся витреальная дегенерация с образованием витреальных шварт, центральный и периферический ретиношизис, пигментная дегенерация, напоминающая пигментный ретинит, осложненные катаракты, гемералопия с отсутствием электроретинограммы, прогрессирующее ухудшение зрительных функций и простой рецессивный тип наследования.Врожденные синдромы являются еще одним источником витреоретинальной патологии и отслоек сетчатки. Среди наиболее часто встречающихся эсенциальных мезодермальных синдромов следует отметить синдром Элерса-Данло (системной эластодистрофии), клинически проявляющийся чрезвычайной мобильностью и растяжимостью кожи (106,109). Случаи отслоек обычно сопровождаются истончением склеры, что затрудняет проведение склеропластических операций. Тип передачи обычно регулярный или нерегулярный аутосомально-доминантный.
Целый ряд клинических ситуаций сопровождается т.н. ангиоидными полосами на глазном дне, впервые описанные Доулинг в 1899 г. (цит.109). Этот патологический синдром часто ассоциируется с псевдоксантомой эластикум (синдром Грюнблада-Страндберга), деформирующим остеитом Паже (синдром Терри), фибриноидной дегенерацией соединительной ткани (синдром Стиллермана) и серповидноклеточной анемией. Офтальмоскопическая картина ангиоидных полос обычно представляет собой разрывы мембраны Бруха.
Прогрессирование заболеваний зачастую приводит к дегенерации желтого пятна типа Кунта-Юниуса. Передача наследственности обычно происходит по рецессивному типу.Довольно распространено сочетание отслойки сетчатки с синдромом Марфана, сопровождающееся двусторонней эктопией хрусталика, арахнодактилией, гипоплазией мышц, повышенной подвижностью суставов, астенопическим хабитусом, кифосколиозом, отсутствием подкожно-жировой клетчатки и аномалиями аорты и клапанов сердца. Отслойка сетчатки при синдроме Марфана зачастую протекает особенно тяжело и сопровождается обычно заворотами разрывов сетчатки, что обычно объясняется отсутствием или только частичной отслойкой задней гиалоидной мембраны. Разрывы сетчатки занимают обычно всю окружность и сопровождаются сокращением фибропластически измененных гиалоидных мембран и выраженной ретракцией сетчатки.
Следствием нарушения развития мезодермальной закладки является т.н. синдром Марчезани, который характеризуется брахиморфией (небольшой рост, короткие пальцы, ограничение подвижности в суставах), сферофакией, микрофакией. Способ передачи обычно промежуточный или доминантный.
Сравнительно нередкими являются также случаи отслойки сетчатки на глазах с колобомами хориоидеи. Разрывы сетчатки, как правило, располагаются по краям колобомы или вблизи от нее.
Из эссенциальных нейроэктодермальных синдромов наиболее часто встречается сочетание отслойки сетчатки с синдромом Гиппеля-Линдау (ретинальный ангиобластоматоз). Заболевание является семейным, передается по доминантному типу примерно у 20% пациентов (цит.109). Офтальмоскопически обычно наблюдается довольно значительное сосудистое образование в сетчатке с подходящими к нему утолщенными, резко извитыми сосудами. Отслойки сетчатки обычно экссудативного типа сопровождаются субретинальными кровоизлияниями. В конечной стадии это заболевание напоминает наружный экссудативный ретинит Коатса.
Наследственные пигментные аномалии, в частности альбинизм, зачастую сопровождаются отслойкой сетчатки и ретиношизисом в молодом возрасте.
Пациенты с альбинизмом, как правило, страдают близорукостью высокой степени в комбинации с абиотрофическими хориоретинальными и витреальными дегенерациями. В случаях альбинотического глазного дна, сочетающегося с отслойкой сетчатки, применение фотокоагуляции, диатермии или криопексии обычно неэффективно в качестве методов лечения.В группу пигментных хориоидальных абиотрофий входит также хориоидальная атрофия и склероз в сочетании с прогрессирующей близорукостью. Пигментный ретинит или пигментная ретинопатия обычно редко сопровождается отслоением сетчатки, возможно за счет глиоза с выраженным интраретинальным отложением пигмента.
Из синдромов, связанных с хромосомными аномалиями, отслойка сетчатки может возникнуть при двусторонних хориоретинальных и витреальных абиотрофиях, которые в сочетании с хромосомными аномалиями наблюдаются у пациентов с синдромом Ульрих Тюрнера и Кляйнфельтера. Первый синдром характеризуется брахиморфией, аплазией половых органов, сексуальным инфантилизмом, соматическими расстройствами (бочкообразная грудь, торчащие уши, монголоидная складка век, косоглазие, птоз, микрокорнеа). При синдроме Кляйнфельтера отслоение сетчатки наблюдается сравнительно редко и возникновение ее обычно сопровождается выраженной дегенерацией и разжижением стекловидного тела, образованием фибропластических мембран в преретинальной зоне витреума. Аутосомные аномалии включают обычно синдром Дауна или монголизм, синдром Пето и синдром Эдварда. Синдромы сопровождаются выраженным образованием преретинальных и периретинальных мембран с фиксированными складками сетчатки без видимых ее разрывов.
Среди вторичных гиалоидоретинопатий наибольшее значение в плане потенциальной отслойки сетчатки имеет ретролентарная фиброплазия, которая встречается у недоношенных детей, находившихся под гипербарической оксигенацией. Возникающая иногда при ретролентарной фиброплазии отслойка сетчатки характеризуется тяжестью течения, гигантскими разрывами и смещением всей сетчатки в нижне-наружный квадрант. Стертые формы ретролентарной фиброплазии представляют целый ряд клинических проявлений, включающих в себя дистрофию сетчатки, серповидную отслойку сетчатки, аномалии сосудов ретины, смещение макулы в височную половину.