1.3.3. бНГПЮЯРМНЕ ХГЛЕМЕМХЕ ЛНПТНЖХРНЛЕРПХВЕЯЙХУ ОНЙЮГЮРЕКЕИ ЛХРНУНМДПХИ
ХГ ЙКЕРНЙ ПЮГКХВМШУ ФХБНРМШУ Х ВЕКНБЕЙЮ ХГСВЕМН МЮХАНКЕЕ ОНДПНАМН Х УНПНЬН НРПЮФЮЕР СЙЮГЮММШЕ БШЬЕ ЯРПСЙРСПМШЕ Х ТСМЙЖХНМЮКЭМШЕ ХУ МЮПСЬЕМХЪ, ОПХВ╦Л МЕЙНРНПШЕ ХЯЯКЕДНБЮРЕКХ НОХЯШБЮЧР, ОН ЯСЫЕЯРБС, НДМХ Х РЕ ФЕ ДЕЯРПСЙРХБМШЕ ХГЛЕМЕМХЪ, ОНКЭГСЪЯЭ ПЮГКХВМШЛХ ЙПХРЕПХЪЛХ ХУ НЖЕМЙХ.
мЮОПХЛЕП, ГЮТХЙЯХПНБЮМШ РЮЙХЕ ДЮММШЕ: ЯМХФЕМХЕ НРМНЬЕМХЪ ДКХМШ БМСРПЕММХУ ЛЕЛАПЮМ Й НАЗ╦ЛС ЙКЕРЙХ, СЙНПНВЕМХЕ ЙПХЯР Х СЛЕМЭЬЕМХЕ ЙНКХВЕЯРБЮ ЛХРНУНМДПХИ (Tate, Herbener, 1976); ЯМХФЕМХЕ НРМНЬЕМХЪ ДКХМШ БМСРПЕММХУ ЛЕЛАПЮМ Й ОКНЫЮДХ Х НАЗ╦ЛС ЛХРНУНМДПХИ, СЛЕМЭЬЕМХЕ Б МХУ ЙПХЯР Х ВХЯКЮ ЯЮЛХУ ЛХРНУНМДПХИ ОПХ СБЕКХВЕМХХ ЯПЕДМЕЦН ХУ ПЮГЛЕПЮ (Eleming et al., 1985); ВЮЯРЭ ЛХРНУНМДПХИ ХЛЕЕР ОПНЯБЕРК╦ММШИ ЛЮРПХЙЯ, ПЮЯЬХПЕММШЕ ЛЕФЙПХЯРМШЕ ОПНЛЕФСРЙХ, ДХЯЙНЛОКЕЙЯХПНБЮММШЕ ЙПХЯРШ; Б НОПЕДЕК╦ММНЛ ОПНЖЕМРЕ ЛХРНУНМ-ДПХИ ЙПХЯРШ Х БМСРПЕММХЕ ЛЕЛАПЮМШ ПЮГПСЬЕМШ, Ю ТНПЛЮ Х ПЮГЛЕПШ ЯЮЛХУ НПЦЮМЕКК ХГЛЕМЕМШ √ МЮПЪДС Я НАШВМШЛХ ОПХЯСРЯРБСЧР ЙПСОМШЕ, Ю ХМНЦДЮ ЦХЦЮМРЯЙХЕ (тПНКЭЙХЯ, яРСОХМЮ, 1982); СЛЕМЭЬЕМХЕ ЙНМРСПМНИ ДКХМШ ЙПХЯР ЛХРНУНМДПХИ Б ПЮЯВ╦РЕ МЮ ОКНЫЮДЭ ЩРХУ НПЦЮМЕКК ХКХ МЮ ОКНЫЮДЭ ЙКЕРЙХ, ЯМХФЕМХЕ ХУ ЙНКХВЕЯРБЮ ОПХ СБЕКХВЕМХХ ПЮГЛЕПНБ НЯРЮБЬХУЯЪ ЛХРНУНМДПХИ (кХРНЬЕМЙН, 1986).оНЪБКЕМХЕ ЛХРНУНМДПХИ АНКЭЬХУ ПЮГЛЕПНБ НАШВМН ПЮЯЯЛЮРПХБЮЧР ЙЮЙ ПЕЮЙЖХЧ ЙНЛОЕМЯЮЖХХ МЮ СЛЕМЭЬЕМХЕ ТНМДЮ МНПЛЮКЭМН ТСМЙЖХНМХПСЧЫХУ НПЦЮМЕКК, МН ДЕИЯРБХРЕКЭМНИ ЙНЛОЕМЯЮЖХХ, ОН-БХДХЛНЛС, МЕ ОПНХЯУНДХР ББХДС ДЕТЕЙРМНЯРХ Х ЩРХУ АНКЭЬХУ ЛХРНУНМДПХИ Б СЯКНБХЪУ ХГАШРНВМНИ ОЕПНЙЯХДЮЖХХ. бНГПЮЯРМНЕ СБЕКХВЕМХЕ ЯПЕДМЕЦН ПЮГЛЕПЮ ЛХРНУНМДПХИ Х ХГЛЕМЕМХЕ ДПСЦХУ ЛНПТНЖХРНЛЕРПХВЕЯЙХУ ОЮПЮЛЕРПНБ ХУ кХРНЬЕМЙН (1985) НАЗЪЯМЪЕР ЯМХФЕМХЕЛ ОПХ ЯРЮПЕМХХ ЯЙНПНЯРХ ЯХМРЕГЮ АЕКЙНБ, ЙНДХПСЕЛШУ ЛХРНУНМДПХЪЛХ, ОПХ ЯНУПЮМЕМХХ ЩРНИ ЯЙНПНЯРХ ДКЪ АЕКЙНБ ЛХРНУНМДПХИ ЪДЕПМНЦН ЙНДХПНБЮМХЪ. рЮЙНИ ДХЯАЮКЮМЯ Б ЯХМРЕГЕ ЛХРНУНМДПХЮКЭМШУ АЕКЙНБ ПЮГМНЦН ЙНДХПНБЮМХЪ НАЗЪЯМЪЕРЯЪ, БНГЛНФМН, РЕЛ, ВРН ОПХ ЯРЮПЕМХХ НПЦЮМХГЛЮ Б Ъдмй БНГПЮЯРЮЕР ОПЕДЯРЮБКЕММНЯРЭ ЛРдмй-ОНДНАМШУ ОНЯКЕДНБЮРЕКЭМНЯРЕИ БЯКЕДЯРБХЕ ХМРЕЦПЮЖХХ ТПЮЦЛЕМРНБ ХКХ ЖЕКШУ ЛНКЕЙСК ЛРдмй Б ЪДЕПМШИ ЦЕМНЛ (лНГФСУХМЮ Х ДП., 1994).
яБН╦ ОПЕДЯРЮБКЕМХЕ ЩРХ ЮБРНПШ ОНДРБЕПДХКХ Б ЩЙЯОЕПХЛЕМРЮУ МЮ ЙПШЯЮУ. б Ъдмй ОЕВЕМХ ЯРЮПШУ ФХБНРМШУ ЯНДЕПФЮМХЕ ЛРдмй-ОНДНАМШУ ОНЯКЕДНБЮРЕКЭМНЯРЕИ БНГПЮЯРЮКН ДН 240% ОН НРМНЬЕМХЧ Й РЮЙНБНЛС С ЛНКНДШУ ЙПШЯ, ОПХВ╦Л
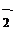 | ||||
| ||||
оНЙЮГЮРЕКЭМН НАНАЫЕМХЕ ЯРПСЙРСПМШУ ОПНЪБКЕМХИ ЯРЮПЕМХЪ ЙКЕРНЙ, НЯСЫЕ-ЯРБК╦ММНЕ яРСОХМНИ Я ЯНЮБР. (1993). хЯОНКЭГСЪ ЛНПТН-ТСМЙЖХНМЮКЭМШИ ЮМЮКХГ БШЪБКЕММШУ БНГПЮЯРМШУ ХГЛЕМЕМХИ Б ЙКЕРЙЮУ ПЮГКХВМШУ НПЦЮМНБ, НМХ ПЮГЦПЮ-МХВХКХ МЮХАНКЕЕ УЮПЮЙРЕПМШЕ ДКЪ ОПНЖЕЯЯЮ ЯРЮПЕМХЪ ЯДБХЦХ МЮ ДЕЯРПСЙРХБМШЕ Х ЮДЮОРХБМШЕ. пЕГСКЭРЮРШ РЮЙНЦН ЙЮВЕЯРБЕММНЦН ЮМЮКХГЮ, ЙЮЯЮЧЫХЕЯЪ, Б ВЮЯРМН-ЯРХ, ДЕЯРПСЙРХБМШУ ХГЛЕМЕМХИ ЛХРНУНМДПХИ, ОПЮЙРХВЕЯЙХ ОНКМНЯРЭЧ ЯНБОЮДЮ-ЧР Я ПЮЯЯЛНРПЕММШЛХ БШЬЕ. дБНИЯРБЕММНЯРЭ ФЕ ОПНЪБКЕМХЪ ОЕПЕЯРПНЕЙ ЛХРН-УНМДПХИ Х ДПСЦХУ БМСРПХЙКЕРНВМШУ ЯРПСЙРСП ОПХ ЯРЮПЕМХХ АШКН АШ КНЦХВМН СБЪГЮРЭ Я СПНБМЕЛ ДХЯАЮКЮМЯЮ ∆ (он-юн) Б ЙКЕРЙЮУ, ЙНРНПШИ ПЮГКХВЕМ МЮ ПЮГМШУ ЩРЮОЮУ ОПНЖЕЯЯЮ ЯРЮПЕМХЪ Х ХМДХБХДСЮКЕМ ДКЪ ЙЮФДНИ ЙКЕРЙХ (Б ЯБЪГХ Я ЦЕРЕПНУПНММНЯРЭЧ ЩРНЦН ОПНЖЕЯЯЮ √ ЯЛ. О. 1.1.2). рЮЙХЛ НАПЮГНЛ, Я БНГПЮЯРНЛ РЙЮМЕБНЕ ДШУЮМХЕ Х ЯЙНПНЯРЭ ОНРПЕАКЕМХЪ н2 ОПХ КЧАНЛ ЯСАЯРПЮРЕ ЯМХФЮЧРЯЪ МЕ РНКЭЙН ХГ-ГЮ МЮПСЬЕМХЪ ПЮАНРШ ДШУЮРЕКЭМНИ ЖЕОХ, МН Х БЯКЕДЯРБХЕ СЛЕМЭ-ЬЕМХЪ ЙНКХВЕЯРБЮ Х, ЯКЕДНБЮРЕКЭМН, НАЫЕИ ╚ЛНЫМНЯРХ╩ ЯЮЛХУ ЛХРНУНМДПХИ.
б ХРНЦЕ ЕЫ╦ АНКЕЕ БНГПЮЯРЮЧР МЕНАУНДХЛШЕ ДКЪ ЯРЮПЕМХЪ БМСРПХЙКЕРНВМШЕ ЦХОЕПНЙЯХЪ Х ОЕПНЙЯХДЮРХБМШИ ЯРПЕЯЯ.лХРНУНМДПХХ ЛНЦСР ДЕЦПЮДХПНБЮРЭ ЙЮЙ ОПХ ЦХОЕПНЙЯХХ, РЮЙ Х ОПХ ДЕТХ-ЖХРЕ н2 БМСРПХ МХУ. б ЩРНИ ЯБЪГХ БЮФМН РЮЙФЕ ПЮГНАПЮРЭЯЪ Я ХГЛЕМЕМХЪЛХ Б ОПНЖЕЯЯЮУ РПЮМЯОНПРЮ н2 Й ЯЮЛХЛ ЙКЕРЙЮЛ. йЮЙ ХГБЕЯРМН (мЮЦНПМШИ Х ДП., 1963), БНГПЮЯРМШЕ ХГЛЕМЕМХЪ ОПНХЯУНДЪР Х Б ЯНЯСДХЯРНИ ЯХЯРЕЛЕ НПЦЮМХГЛЮ. нЯНАЕММН ГМЮВХРЕКЭМШ НМХ Б ЮПРЕПХЪУ Х ЮПРЕПХНКЮУ: ЯРЕМЙХ ХУ Б НМРНЦЕМЕГЕ ОНЯРЕОЕММН РЕПЪЧР ЩКЮЯРХВМНЯРЭ, ЯОНЯНАМНЯРЭ ЯНЯСДНБ ХГЛЕМЪРЭ ЯБНИ ОПНЯБЕР СЛЕМЭЬЮЕРЯЪ, Б ХУ РЙЮМЪУ МЮЯРСОЮЧР ЯЙКЕПНРХВЕЯЙХЕ ХГЛЕМЕМХЪ, НАЫЕЕ ПСЯКН ЙЮОХККЪПНБ Х ЮПЕРПХНК ЯСФЮЕРЯЪ, Ю ЙПНБЪМНЕ ДЮБКЕМХЕ Б ЯХЯРЕЛЕ СБЕКХВХБЮЕРЯЪ. щРХ ХГЛЕ-МЕМХЪ БЛЕЯРЕ ЯН ЯРЮПЕМХЕЛ ЩПХРПНЖХРНБ Х СУСДЬЕМХЕЛ ТСМЙЖХХ БМЕЬМЕЦН ДШУЮМХЪ, ЕЯРЕЯРБЕММН, ЯОНЯНАЯРБСЕР ГЮРСУЮЧЫЕЛС Б НМРНЦЕМЕГЕ ОНРПЕАКЕМХЧ н2 РЙЮМЪЛХ НПЦЮМХГЛЮ. б НОХЯШБЮЕЛНИ ЯХРСЮЖХХ ДНЯРЮБЙЮ н2 Б ЙКЕРЙХ МЕЙНРНПШУ РЙЮМЕИ Б ЕДХМХЖС БПЕЛЕМХ ЛНФЕР НЙЮГЮРЭЯЪ ЛЕМЭЬЕ ТХГХНКНЦХВЕЯЙХ МЕНАУН-ДХЛНЦН ЕЦН ЙНКХВЕЯРБЮ. пЕЮЙЖХЪ РЮЙХУ ЙКЕРНЙ МЮ ДЕТХЖХР н2, ЯЙНПЕЕ БЯЕЦН, АСДЕР НДМНГМЮВМНИ: СЛЕМЭЬЕМХЕ ЙНКХВЕЯРБЮ ДШУЮРЕКЭМШУ ТЕПЛЕМРНБ Б ЯННР-БЕРЯРБХХ Я н2-ГЮБХЯХЛШЛ ЛЕУЮМХГЛНЛ ПЕЦСКЪЖХХ ХУ ЯНДЕПФЮМХЪ (Murphy et al., 1984) Х НАЫЕИ ╚ЛНЫМНЯРХ╩ ЛХРНУНМДПХИ, Р.Е. ЮДЮОРЮЖХЪ Й СЯКНБХЪЛ ЙХЯКН-ПНДМНЦН ЦНКНДЮМХЪ. дЕЯРПСЙЖХЪ ╚КХЬМХУ╩ ЛХРНУНМДПХИ Б СЯКНБХЪУ ЦХОНЙЯХХ Х ОЮДЕМХЕ НЙХЯКХРЕКЭМНЦН ТНЯТНПХКХПНБЮМХЪ ЛНЦСР, Б ВЮЯРМНЯРХ, ОПНУНДХРЭ ОН ЯУЕЛЕ (яНПНЙНБНИ, бКЮДХЛХПНБ, 1975): БШУНД яЮ2+ ХГ ЛЮРПХЙЯЮ → ЮЙРХБЮЖХЪ ТНЯТНКХОЮГШ ю2 ХНМЮЛХ ЙЮКЭЖХЪ → ДЕГШМРЕЦПЮЖХЪ ЛЕЛАПЮМ ЛХРНУНМДПХИ → МЮПСЬЕМХЕ ХУ ЯРПСЙРСПШ Х ТСМЙЖХИ.
рНМЙНЯРХ н2-ГЮБХЯЛНЦН ЛЕУЮМХГЛЮ ПЕЦСКЪЖХХ ЙНКХВЕЯРБЮ ДШУЮРЕКЭМШУ ТЕПЛЕМРНБ ОНЙЮ ЕЫ╦ МЕ СЯРЮМНБКЕМШ. оНЩРНЛС ХМРЕПЕЯМЮ ПЮАНРЮ, ЦДЕ ОСР╦Л ЯНГДЮМХЪ ХМЦХАХРНПНЛ ДШУЮМХЪ ЛХРНУНМДПХЮКЭМНЦН ЯРПЕЯЯЮ ХГСВЮКХ ЯБЪГЭ ЛЕФДС ЛХРНУНМДПХЪЛХ Х ЪДПНЛ Б ЙКЕРЙЮУ ВЕКНБЕЙЮ. нЙЮГЮКНЯЭ, ВРН ОПХ ЯМХ-ФЕМХХ ДШУЮМХЪ ЛХРНУНМДПХИ ДН 50-70 % НР МНПЛШ Б ЙКЕРЙЮУ МЮАКЧДЮЕРЯЪ ЮЙРХБЮЖХЪ ЩЙЯОПЕЯЯХХ ЖХРНУПНЛНБ b1 Х c, ЙНРНПЮЪ НОНЯПЕДНБЮМЮ ОНБШЬЕМХЕЛ СПНБМЪ м2н2 Б ЙКЕРЙЮУ Х ЯОНЯНАЯРБСЕР ОПНЖЕЯЯС ХУ БШФХБЮМХЪ.
оПХ АНКЕЕ БШПЮФЕММНЛ ХМЦХАХПНБЮМХХ ДШУЮМХЪ (ДН 40% НР МНПЛШ) Х ЯННРБЕРЯРБЕММН АНКЕЕ ХМРЕМЯХБМНЛ БНГПЮЯРЮМХХ ЯНДЕПФЮМХЪ м2н2 ЪДЕПМШЕ ЦЕМШ СЙЮГЮММШУ ЖХРНУПНЛНБ МЕ ЮЙРХБХПСЧРЯЪ (Suzuki H. et al., 1998). дЮММШЕ ТЮЙРШ ДЕЛНМЯРПХПСЧР ЯСЫЕЯРБНБЮМХЕ, ОН ЙПЮИМЕИ ЛЕПЕ, РП╦У БГЮХЛНЯБЪГЮММШУ ОПХМЖХОХЮКЭМШУ ЩТТЕЙРНБ: ХМЦХАХПНБЮМХЕ ЛХРНУНМДПХЮКЭМНЦН ДШУЮМХЪ БЕД╦Р Й БМСРПХЙКЕРНВМНИ ЦХОЕПНЙЯХХ Х ЯННРБЕРЯРБЕММН Й ХГАШРНВМНЯРХ СПНБМЪ ютй; ОНЯКЕДМХЕ Х, Б ВЮЯРМНЯРХ, м2н2 ОПХБКЕВЕМШ Й ЩЙЯОПЕЯЯХХ ПЮГКХВМШУ ЪДЕПМШУ ЦЕМНБ, Б РНЛ ВХЯКЕ ЙНДХПСЧЫХУ ДШУЮРЕКЭМШЕ ТЕПЛЕМРШ b1 Х c ДКЪ ЛХРНУНМДПХИ; ЩРХ ОПНЪБКЕМХЪ ВЮЯРХВМН ЙНЛОЕМЯХПСЧР МЮПСЬЕМХЪ (МЕДНЯРЮРНВМНЯРЭ) ЛХРНУНМДПХЮКЭМНЦН ДШУЮМХЪ, ЕЯКХ НМХ МЕ ЦКСАНЙХЕ.йЯРЮРХ, БЮФМН НРЛЕРХРЭ ГДЕЯЭ, ВРН ЛЕУЮМХГЛ ПЕЦСКЪЖХХ ЩЙЯОПЕЯЯХХ ЦЕМНБ ДШУЮРЕКЭМШУ ТЕПЛЕМРНБ ЙНМЖЕМРПЮЖХЕИ н2 ╚БМЕДП╦М╩ СФЕ МЮ СПНБМЕ ДПНФФЕИ. оНЙЮГЮМЮ, МЮОПХЛЕП, ГЮБХЯХЛНЯРЭ ЩЙЯОПЕЯЯХХ МЕЯЙНКЭЙХУ ЪДЕПМШУ ЦЕМНБ Saccharomyces cerevisiae, ЙНДХПСЧЫХУ АЕКЙХ РЕПЛХМЮКЭМНИ ВЮЯРХ ДШУЮРЕКЭМНИ ЖЕОХ (ЖХРНУПНЛ Я, ЖХРНУПНЛ-Я-НЙЯХДЮГС), ХЛЕММН НР ЙНМЖЕМРПЮЖХХ н2, Ю МЕ ОПНЯРН ОПХЯСРЯРБХЪ ХКХ НРЯСРЯРБХЪ н2 (Burke et al., 1997). нЙЮГЮКНЯЭ, ВРН ЩЙЯОПЕЯЯХЪ МЕЙНРНПШУ ЦЕМНБ (яну4, яну √ яну9) ОНЯРЕОЕММН ЯМХФЮЕРЯЪ ОПХ ХГЛЕМЕМХХ ЙНМЖЕМРПЮЖХХ н2 НР 200 ЛЙл (БНГДСУ) ДН ОНПНЦНБНИ, ЙНРНПЮЪ ДКЪ ЩРХУ ЦЕМНБ НВЕМЭ МХГЙЮ (0.5 √ 1.0 ЛЙл). мХФЕ ЙХЯКНПНДМНЦН ОНПНЦЮ ЩЙЯОПЕЯЯХЪ ПЕГЙН ОЮДЮЕР. оПЮБДЮ, Б ЩРНИ ФЕ ПЮАНРЕ ОНЙЮГЮМН ЯСЫЕЯРБНБЮМХЕ Х ╚ЦХОНЙЯХВЕЯЙХУ╩ ЦЕМНБ (яну5Б, CYC7), ЩЙЯОПЕЯЯХПСЧЫХУЯЪ РНКЭЙН ОПХ ОЮДЕМХХ ЙНМЖЕМРПЮЖХХ н2 МХФЕ 0.5 ЛЙл, Ю РЮЙФЕ ТЕМНЛЕМЮ ЙНЩЙЯОПЕЯЯХХ Б НОПЕДЕК╦ММНЛ ХМРЕПБЮКЕ ЙНМЖЕМРПЮЖХХ н2 ╚ЮЩПНАМШУ╩ Х ╚ЦХОНЙЯХВЕЯЙХУ╩ ЦЕМНБ, ЙНДХПСЧЫХУ ЯННРБЕРЯРБСЧЫХЕ ХЛ ХГНТНПЛШ ЖХРНУПНЛЮ Я Х ЖХРНУПНЛ-Я-НЙЯХДЮГШ.
оПХЛЕВЮРЕКЭМШЛ РЮЙФЕ ОПЕДЯРЮБКЪЕРЯЪ ДПСЦНИ ТЮЙР: НАМЮПСФЕМХЕ СФЕ МЮ СПНБМЕ ОПНЯРЕИЬХУ НДМНЙКЕРНВМШУ НПЦЮМХГЛНБ, Б ВЮЯРМНЯРХ С Dictyostelium, ЙХЯКНПНДМНЦН ОЕПЕЙКЧВЮРЕКЪ РПЮМЯЙПХОЖХХ. пЕВЭ ХД╦Р Н РНЛ, ВРН ЛЮКЮЪ ЯСАЗЕДХМХЖЮ ЖХРНУПНЛ-Я-НЙЯХДЮГШ ХЛЕЕР ДБЕ БГЮХЛННАЛЕМХБЮЕЛШЕ ХГНТНПЛШ, ОНЪБКЕМХЕ ЙНРНПШУ ПЕЦСКХПСЕРЯЪ н2 Б ГЮБХЯХЛНЯРХ НР СЯКНБХИ ПНЯРЮ (Б РНЛВХЯКЕ, ОН-БХДХЛНЛС, Х НР ЙНМЖЕМРПЮЖХХ н2).
щРХ ЯСАЗЕДХМХЖШ ЙНДХПСЧРЯЪ ЯНЯЕДМХЛХ ЦЕМЮЛХ, ЯБЪГЮММШЛХ РЮМДЕЛМН ╚ЦНКНБЮ √ УБНЯР╩ Х ПЮГДЕК╦ММШЛХ ЙНПНРЙХЛ ЛЕФЦЕММШЛ ЯЕМЛЕМРНЛ, ЙНРНПШИ Х БШОНКМЪЕР ПНКЭ ЙХЯКНПНДМНЦН ОЕПЕЙКЧВЮРЕКЪ РПЮМЯЙПХОЖХХ. рЮЙЮЪ ЯРПСЙРСПМЮЪ НПЦЮМХГЮЖХЪ Х ПЮГКХВМЮЪ ЯРЮАХКЭМНЯРЭ ХГНТНПЛ ЯСАЗЕДХМХЖШ ТЕПЛЕМРЮ НАЗЪЯМЪЧР МЕНАШВМСЧ ВСБЯРБХРЕКЭМНЯРЭ Й н2 ОЕПЕЙКЧВЮЧЫЕЦН ЛЕУЮМХГЛЮ (Bisson et al., 1997). оНЯКЕДМХИ ОПЕДЯРЮБКЪЕР, ОН ЯСЫЕЯРБС, ПЕДЙХИ Б ФХБНИ ОПХПНДЕ ОПХЛЕП ЯХЯРЕЛШ СОПЮБКЕМХЪ Я ОЕПЕЛЕММНИ ЯРПСЙРСПНИ.бНГБПЮЫЮЪЯЭ Й БНОПНЯС Н ЯРЮПЕМХХ ЙКЕРЙХ, НРЛЕРХЛ, ВРН НЦПЮМХВЕМХЕ ОН СЙЮГЮММШЛ БШЬЕ ОПХВХМЮЛ ОНЯРСОКЕМХЪ н2 Й ЙКЕРЙЮЛ Х ЯННРБЕРЯРБЕММН ЯМХФЕМХЕ ЕЦН ОНРПЕАКЕМХЪ ХЛХ ЛНФЕР БМНБЭ ОНБШЯХРЭ БМСРПХЙКЕРНВМНЕ Пн2, Р.Е. ЯНГ-ДЮРЭ СЯКНБХЪ ДКЪ ОНДДЕПФЮМХЪ ДНЯРЮРНВМНЦН ОН БЕКХВХМЕ ДХЯАЮКЮМЯЮ ∆ (он √ юн) Х ОПНДНКФЕМХЪ ОПНЖЕЯЯЮ ЯРЮПЕМХЪ ЙКЕРНЙ. рЮЙНЕ ЯНЯРНЪМХЕ ДНКФМН АШРЭ УЮПЮЙРЕПМШЛ Б НЯМНБМНЛ ДКЪ ОНГДМЕЦН НМРНЦЕМЕГЮ. оПХ ЩРНЛ МЮ МЕЙНРНПНЛ ЩРЮОЕ ЯРЮПЕМХЪ ЛНФЕР НАПЮГНБЮРЭЯЪ ЯХРСЮЖХЪ, ЙНЦДЮ БМНБЭ БНГПНЯЬХЕ ГМЮВЕМХЪ ДХЯАЮКЮМЯЮ ∆ (он √ юн) НЙЮФСРЯЪ ЯННРБЕРЯРБСЧЫХЛХ ДХЮОЮГНМС ХГЛЕМЕМХЪ ∆о (он √ юн). рНЦДЮ Б ОПХМЖХОЕ ЛНФЕР БНГМХЙМСРЭ ЯНЯРНЪМХЕ НАМНБКЕМХЪ РЙЮМЕИ Б ЯРЮПНЛ НПЦЮМХГЛЕ (ЯЛ. О.1.6.1) √ ТЕМНЛЕМ, ЙЮФСЫХИЯЪ ОЮПЮДНЙЯЮКЭМШЛ Х Б РН ФЕ БПЕЛЪ БЕЯЭЛЮ ОПХБКЕЙЮРЕКЭМШЛ ДКЪ ОНМХЛЮМХЪ ЛЕУЮМХГЛЮ НЛНКНФЕМХЪ ПЪДЮ ЯХЯРЕЛ НПЦЮМХГЛЮ Б ОНГДМЕЛ НМРНЦЕМЕГЕ.
йЮЙ ЙНЯБЕММНЕ ЯБХДЕРЕКЭЯРБН МЕДНЯРЮРНВМНЯРХ ЛХРНУНМДПХЮКЭМНЦН ДШУЮМХЪ НАПЮЫЮЧР МЮ ЯЕАЪ БМХЛЮМХЕ Х ТЮЙРШ ЛЮЯЯХБМНИ ОПНДСЙЖХХ ЯБНАНДМШУ ПЮДХЙЮКНБ н2 ЯРЮПЕЧЫХЛХ ЙКЕРЙЮЛХ (ЦЕОЮРНЖХРЮЛХ) ОПХ ПЕНЙЯХЦЕМЮЖХХ ОНЯКЕ ОПЕАШБЮМХЪ ХУ Б ЯНЯРНЪМХХ ЮМНЙЯХХ (De Notaris et al., 1995). дКЪ НАЗЪЯМЕМХЪ ДЮММНЦН ЩТТЕЙРЮ ОПЕДЯРЮБКЪЕРЯЪ ЮПЦСЛЕМРХПНБЮММШЛ ЯКЕДСЧЫЕЕ МЮЬЕ ЯСФДЕМХЕ. оПХ ПЕОЕПТСГХХ ЦЕОЮРНЖХРШ ЛНКНДШУ ЙПШЯ Я ЛЕМЭЬХЛХ ОНРЕПЪЛХ ОЕПЕМНЯЪР ПЕГЙНЕ БНГПЮЯРЮМХЕ ДХЯАЮКЮМЯЮ ∆ (он √ юн), ОНЯЙНКЭЙС ХУ МЕОНБПЕФД╦ММШЕ ЕЫ╦ ЯРЮПЕМХЕЛ ЛХРНУНМДПХХ, ЪБКЪЪЯЭ ЦКЮБМШЛ ЮМРХЙХЯКНПНДМШЛ ТЮЙРНПНЛ Б ЙКЕРЙЕ, АНКЕЕ ХКХ ЛЕМЕЕ ЩТТЕЙРХБМН ОНРПЕАКЪЧР н2 Х НРВЮЯРХ МЕИРПЮКХГСЧР ЯЙЮВЙННАПЮГМНЕ ОНБШЬЕМХЕ Пн2.
б АНКЕЕ ФЕ ЯРЮПШУ ЙКЕРЙЮУ СФЕ ВЮЯРХВМН ДЕТЕЙРМШЕ ЛХРНУНМДПХХ КХЬЕМШ БНГЛНФМНЯРХ НЫСРХЛН БКХЪРЭ МЮ ЯМХФЕМХЕ СПНБМЕИ Пн2 Х онк. рЮЙХЕ ЦХОЕПНЙЯХВЕЯЙХЕ ХГМСРПХ ЙКЕРЙХ ХМРЕМЯХБМЕЕ ОПНДСЖХПСЧР ютй, РЮЙ ЙЮЙ ДЮБМН ХГБЕЯРМН: ЯЙНПНЯРЭ НАПЮГНБЮМХЪ ютй Б УНДЕ ЛМНЦХУ ТЕПЛЕМРЮРХБМШУ Х МЕ ТЕПЛЕМРЮРХБМШУ ПЕЮЙЖХИ Х ЯЮЛННЙХЯКЕМХЪ ПЮГКХВМШУ ЯНЕДХМЕМХИ ОПЪЛН ГЮБХЯХР НР Пн2 (Fridovich, 1975). х ЕЫ╦. оН МЮЬЕЛС ОПЕДЯРЮБКЕМХЧ, ВЕЛ ДНКЭЬЕ ЙКЕРЙХ ЛНКНДШУ ФХБНРМШУ АСДСР МЮУНДХРЭЯЪ Б СЯКНБХЪУ ЮМНЙЯХХ, РЕЛ ОНКМЕЕ Х МЮД╦ФМЕЕ НМХ ЮДЮОРХПСЧРЯЪ Й МЕИ, ЯНЙПЮЫЮЪ, ОПЕФДЕ БЯЕЦН, ВХЯКН ЯБНХУ ЛХРНУНМДПХИ (ЯЛ. БШЬЕ), МН ОПХ ОНЯКЕДСЧЫЕИ ПЕНЙЯХЦЕМЮЖХХ НРБЕРМЮЪ ютй-ЦЕМЕПХПСЧЫЮЪ ПЕЮЙЖХЪ ХУ АСДЕР ОПХАКХФЮРЭЯЪ Й РЮЙНБНИ ЯРЮПЕЧЫХУ ЙКЕРНЙ. оНДПНАМЕЕ ЛЕУЮМХГЛ ПЕОЕПТСГХНММНЦН (ПЕНЙЯХЦЕМЮЖХНММНЦН) ОНПЮФЕМХЪ ЙКЕРНЙ БННАЫЕ НАЯСФДЮЕРЯЪ МЮЛХ Б ЦКЮБЕ 4 (О. 4.2.3).яНБЕПЬЕММН МНБШИ Х, ОН-БХДХЛНЛС, ДНОНКМХРЕКЭМШИ ЙЮМЮК ДКЪ СЯХКЕМХЪ НЙХЯКХРЕКЭМНЦН ЯРПЕЯЯЮ Б ЯРЮПЕЧЫХУ ЙКЕРЙЮУ Б ОПХМЖХОЕ БНГЛНФЕМ, ЕЯКХ ОПХМЪРЭ БН БМХЛЮМХЕ ДЮММШЕ ЯКЕДСЧЫЕЦН МЮАКЧДЕМХЪ. мЮ БРНПНИ ЦНД ФХГМХ С ЙПШЯ ОПНХЯУНДХР ЯМХФЕМХЕ ЮЙРХБМНЯРХ ОЕПЕМНЯВХЙЮ ОХПСБЮРЮ Х ЯННРБЕРЯРБЕММН ЯЙНПНЯРХ ОНДДЕПФХБЮЕЛНЦН ОХПСБЮРНЛ ДШУЮМХЪ Б ЛХРНУНМДПХЪУ ЯЕПДЖЮ. б ЩРХУ НПЦЮМЕККЮУ БШЪБКЕМН РЮЙФЕ СЛЕМЭЬЕМХЕ ~40 % ЯНДЕПФЮМХЪ ЙЮПДХНКХОХМЮ. бБЕДЕМХЕ РЮЙХЛ ЙПШЯЮЛ ЮЖЕРХК-L-ЙЮПМХРХМЮ ОНВРХ ОНКМНЯРЭЧ БНЯЯРЮМЮБКХБЮКН ЙЮЙ РПЮМЯОНПР ОХПСБЮРЮ Х ЯЙНПНЯРЭ ДШУЮМХЪ ДН СПНБМЪ ЛНКНДШУ ЙНМРПНКЭМШУ ЙПШЯ, РЮЙ Х ЙНППЕКХПНБЮБЬЕЕ Я ЩРХЛХ ХГЛЕМЕМХЪЛХ ЯНДЕПФЮМХЕ ЙЮПДХНКХОХМЮ (Paragies et al., 1999). кНЦХЙЮ ЯБЪГХ ЛЕФДС ЩРХЛХ ЩТТЕЙРЮЛХ, МЮ ОЕПБШИ БГЦКЪД, ЙЮФЕРЯЪ МЕ ЯНБЯЕЛ ОНМЪРМНИ. нОХПЮЪЯЭ, НДМЮЙН, МЮ ╚ЙХЯКНПНДМН-ОЕПЕЙХЯМШЕ╩ ОПЕДЯРЮБКЕМХЪ Н ЛЕУЮМХГЛЕ ЯРЮПЕМХЪ ЛНФМН ДНОСЯРХРЭ ПЕЮКХГЮЖХЧ МЕЙНРНПШУ ОПНЖЕЯЯНБ, НАСЯКНБКЕММШУ МЕДНЯРЮРНВМНЯРЭЧ ОХПСБЮРЮ Б ЛЮРПХЙЯЕ ЛХРНУНМДПХИ Х ПЕЮЙЖХИ ЖХЙКЮ РПХЙЮПАНМНБШУ ЙХЯКНР. мЮХАНКЕЕ ЯСЫЕЯРБЕММНИ ДНКФМЮ АШРЭ ОНЯКЕДНБЮРЕКЭМНЯРЭ ХГЛЕМЕМХИ: ЯМХФЕМХЕ ЛХРНУНМДПХЮКЭМНЦН ДШУЮМХЪ (ОНРПЕАКЕМХЪ н2) → БМСРПХЙКЕРНВМЮЪ ЦХОЕПНЙЯХЪ → СЯХКЕМХЕ онк ЛЕЛАПЮМ, ОПЕФДЕ БЯЕЦН ЛЕЛАПЮМ ЛХРНУНМДПХИ → ОЮДЕМХЕ Б МХУ СПНБМЪ КЕЦЙННЙХЯКЪЕЛНЦН ЙЮПДХНКХОХМЮ. оНЯКЕДМЕЕ, Б ЯБНЧ НВЕПЕДЭ, ЯНГДЮЕР ОНКНФХРЕКЭМСЧ НАПЮРМСЧ ЯБЪГЭ ОН ОНДДЕПФЮМХЧ Б ЯРЮПЕЧЫХУ Х ЯРЮПШУ ЙКЕРЙЮУ (Б ДЮММНЛ ЯКСВЮЕ ЯЕПДЖЮ ЙПШЯ) НЯКЮАКЕММНЦН ДШУЮМХЪ Х ЯБНАНДМНПЮДХЙЮКЭМНЦН НЙХЯКХРЕКЭМНЦН ЯРПЕЯЯЮ.
хГ ЖХРХПНБЮММШУ БШЬЕ Х ПЪДЮ ДПСЦХУ ПЮАНР (Alemany et al., 1988; Kim et al., 1988) ЯКЕДСЕР, ВРН ЩМЕПЦННАЕЯОЕВЕММНЯРЭ ЯРЮПЕЧЫХУ ЙКЕРНЙ Х РЙЮМЕИ МЕДНЯРЮРНВМЮ ХГ-ГЮ ЯМХФЕМХЪ ЮЙРХБМНЯРХ ЛХРНУНМДПХИ Х (ХКХ) НЦПЮМХВЕММНЯРХ ВХЯКЮ ОНКМНЖЕММШУ ЩРХУ НПЮМЕКК. дЕТХЖХР юрп ДНКФЕМ ЯННРБЕРЯРБСЧЫХЛ НАПЮГНЛ НРПЮГХРЭЯЪ МЮ АХНЯХМРЕРХВЕЯЙХУ ОПНЖЕЯЯЮУ, ХЯОНКМЕМХХ ПЮГКХВМШУ ТХГХНКНЦХВЕЯЙХУ ТСМЙЖХИ. рЮЙ, ЮДЮОРХБМНЕ СЯХКЕМХЕ ЦКХЙНКХГЮ Б ЯБЪГХ ЯН ЯМХФЕМХЕЛ ЯНДЕПФЮМХЪ юрп Б ЯРЮПНЯРХ НАЗЪЯМЪЕРЯЪ НЯКЮАКЕМХЕЛ ЩТТЕЙРЮ оЮЯРЕПЮ. б НЯМНБЕ ОНЯКЕДМЕЦН КЕФЮР ЛЕУЮМХГЛШ ЮККНЯРЕПХВЕЯЙНЦН ХМЦХАХПНБЮМХЪ ЦЕЙЯНЙХМЮГШ ЦКЧЙНГН-6-ТНЯТЮРНЛ Х ТНЯТНТПСЙРНЙХМЮГШ ОПНДСЙРНЛ юрп, ОНЩРНЛС ОПХ ДЕТХЖХРЕ юрп ЮЙРХБМНЯРЭ СЙЮГЮММШУ ТЕПЛЕМРНБ ОНБШЬЮЕРЯЪ (аНЦЮЖЙЮЪ, 1968). дПСЦНИ ОПХЛЕП √ МЮПСЬЕМХЕ Б ОПНЖЕЯЯЕ ЯРЮПЕМХЪ ТСМЙЖХНМХПНБЮМХЪ МЕИПНМНБ ХГ-ГЮ ЯМХФЕМХЪ ВХЯКЮ ЮЙРХБМН ДЕИЯРБСЧЫХУ ЛХРНУНМДПХИ Х, ЯКЕДНБЮРЕКЭМН, МЕДНЯРЮРНВМНЯРХ РПЕАСЕЛНИ ХЛ ЩМЕПЦХХ. рЮЙНИ ТЮЙР СЯРЮМНБКЕМ, Б ВЮЯРМНЯРХ, ОПХ ХЯЯКЕДНБЮМХХ ЛНПТНТСМЙЖХНМЮКЭМШУ ХГЛЕМЕМХИ Б ЛХРНУНМДПХЪУ ЙКЕРНЙ оСПЙХМЕ ЛНГФЕВЙЮ ЙПШЯ Wistar 3-, 12- Х 24-ЛЕЯЪВМНЦН БНГПЮЯРЮ (Fattoretti et al., 1996). мН ЦКЮБМШЕ МЕЦЮРХБМШЕ ОНЯКЕДЯРБХЪ ДНКФМШ НОПЕДЕКЪРЭЯЪ МЕ ДЕТХЖХРНЛ юрп Х Яюлп, Ю, ЯЙНПЕЕ БЯЕЦН, БНГМХЙЮЧЫХЛ ОПХ ЯРЮПЕМХХ ЙКЕРНЙ ЯНЯРНЪМХЕЛ ОЕПНЙЯХЦЕМЮЖХХ. мЮОПХЛЕП, ЯМХФЕМХЕ ЩМЕПЦЕРХВЕЯЙНИ ЮЙРХБМНЯРХ ЛХРНУНМДПХИ Б ЙКЕРЙЮУ ЛНВЕБНЦН ОСГШПЪ ЙПШЯ 30-ЛЕЯЪВМНЦН БНГПЮЯРЮ ЛНФЕР ОПХБНДХРЭ Й СУСДЬЕМХЧ ТСМЙЖХНМЮКЭМШУ БНГЛНФМНЯРЕИ БЯЕЦН НПЦЮМЮ БЯКЕДЯРБХЕ РНЦН, ВРН ОНБШЬЮЕРЯЪ БЕПНЪРМНЯРЭ ОНБПЕФДЕМХЪ ЕЦН РЙЮМЕИ ХГАШРНВМШЛХ ютй (Tong-Long et al., 1998).
еЫЕ ОН РЕЛЕ 1.3.3. бНГПЮЯРМНЕ ХГЛЕМЕМХЕ ЛНПТНЖХРНЛЕРПХВЕЯЙХУ ОНЙЮГЮРЕКЕИ ЛХРНУНМДПХИ:
- нЯНАЕММНЯРХ БНГПЮЯРМШУ ХГЛЕМЕМХИ МЕЙНРНПШУ АХНУХЛХВЕЯЙХУ ОНЙЮГЮРЕКЕИ ЙПНБХ
- 3.1. бНГПЮЯРМШЕ ХГЛЕМЕМХЪ ПЕТПЮЙЖХХ
- бНГПЮЯРМШЕ ХГЛЕМЕМХЪ Б ЯХМЮОЯЮУ
- 1.3 бНГПЮЯРМШЕ ХГЛЕМЕМХЪ ЯКХГХЯРНИ НАНКНВЙХ ФЕКСДЙЮ
- 4. бНГПЮЯРМЮЪ ЯРПСЙРСПЮ ГЮАНКЕБЮЕЛНЯРХДЮЕРЯЪ Б ЩЙЯРЕМЯХБМШУ Х ХМРЕМЯХБМШУ ОНЙЮГЮРЕКЪУ.
- 1.4. бНГПЮЯРМШЕ ХГЛЕМЕМХЪ ДПСЦХУ ЙКЕРНВМШУЯРПСЙРСП Х ТСМЙЖХИ
- бНГПЮЯРМШЕ ХГЛЕМЕМХЪ ПЕЯМХВМНЦН ОНЪЯЙЮ.
- 22. бНГПЮЯРМШЕ ХГЛЕМЕМХЪ Б МЮЯКЕДСЕЛНЯРХ ХМРЕККЕЙРЮ Х НРДЕКЭМШУ ЙНЦМХРХБМШУ ОПНЖЕЯЯНБ.
- оНЙЮГЮРЕКХ ЙЮВЕЯРБЮ ФХГМХ ДЕРЕИ ПЮГКХВМШУ БНГПЮЯРМШУ ЦПСОО
- бНГПЮЯРМЮЪ ДХМЮЛХЙЮ ХГЛЕМЕМХИ ЩМДНРЕКХЪ БХГСЮКЭМН МЕХГЛЕМЕММШУ ЯНЯСДНБ
- хГЛЕМЕМХЪ ОНЙЮГЮРЕКЕИ ОЕПХТЕПХВЕЯЙНИ ЙПНБХ.
- хГЛЕМЕМХЪ ОНЙЮГЮРЕКЕИ ЯХЯРЕЛШ ЦЕЛНЯРЮГЮ.
- хГЛЕМЕМХЪ АХНУХЛХВЕЯЙХУ ОНЙЮГЮРЕКЕИ.
- хГЛЕМЕМХЕ АХНУХЛХВЕЯЙХУ ОНЙЮГЮРЕКЕИ.