Изменения в организме при стрессе
В настоящее время показано, что стресс сопровождается функциональными (нейроэндокринными, обменными) и морфологическими изменениями. Доказана роль стресса как главного этиологического фактора язвенных поражений слизистой желудка, гипертонической болезни, атеросклероза, нарушений структуры и функции сердца, формирования иммунодефицитных состояний и злокачественных опухолей, нарушений обмена веществ (рис 9.1).
Патогенез язв желудка при стрессе. Язвы желудка образуются как обязательный признак первой стадии стресс-реакции. У человека формирование язв наблюдается при стрессе, вызванном конфликтом между необходимостью осуществлять пищевую, половую, оборонительные реакции и запретом или невозможностью их осуществления. У животных аналогичная ситуация моделируется при формалиновом стрессе, иммобилизации, болевом раздражении и невозможностью животных уйти от болевого воздействия. Язвы желудка и кишечника сейчас обнаружены практически при всех сильных стрессорных воздействиях, а у человека особенно после сильных эмоциональных переживаний.
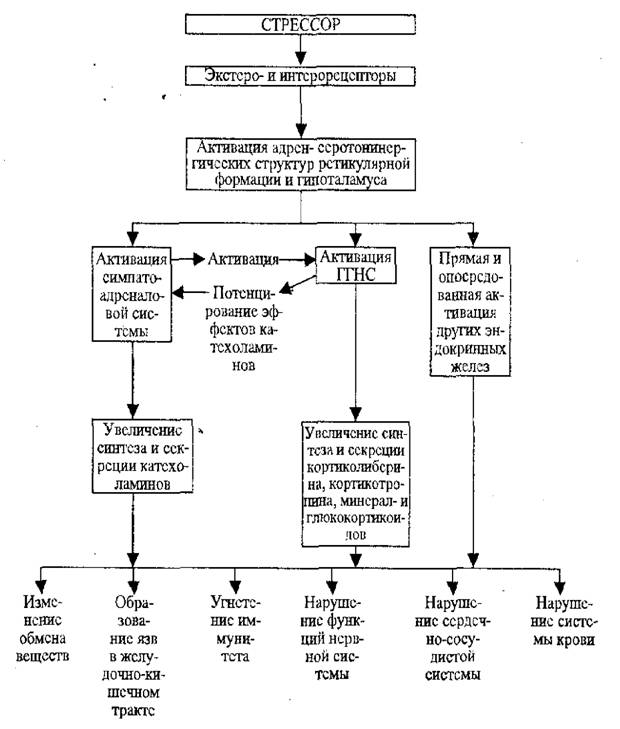
Рис. 9.1. Общий патогенез стресса
Показано, что язвы желудка и кишечника развиваются не во время самого стрессорного воздействия, а спустя некоторое время (в эксперименте, обычно на голодных животных). Полагают, что в результате возбуждения симпато-адреналовой системы возникает спазм
артериол мышечной оболочки желудка, стаз крови, повышение проницаемости сосудов, кровоизлияния и некроз. Одновременно подавляется секреция желудочного сока. Лишь после прекращения стрессорного воздействия восстанавливается, а затем повышается активность парасимпатического отдела нервной системы и усиливается секреция желудочного сока. Ишемизированные и некротизированные участки слизистой оболочки подвергаются перевариванию с образованием язв (Ф.З.
Меерсон).Таким образом, сильное возбуждение симпато-адреналовой системы при стрессе вызывает повреждение слизистой желудка, а последующее повышение парасимпатических влияний и усиление секреции желудочного сока приводят к формированию язв.
Нарушения сердечно-сосудистой системы при стрессе. Активация при стрессе симпато-адреналовой системы вызывает учащение ритма сердечных сокращений, увеличение систолического и минутного объема кровообращения, общего периферического сопротивления, следствием которых является подъем системного артериального давления.
При длительном и интенсивном стрессе регистрируется повреждение миокарда, основными причинами которого являются высокие концентрации катехоламинов, активирующие перекисное окисление липидов, а образующиеся в результате этого гидроперекиси повреждают биомембраны клеток сердца и других органов и тканей (мышц, аорты). По данным Ф.З. Меерсона, перекисное окисление липидов для различных органов при стрессе сохраняется от 2 до 5 суток. Повышение проницаемости мембран лизосом кардиомиоцитов и выход протеолитических ферментов в цитоплазму и кровь вызывает более значительное повреждение мембран клеток. Очаговые контрактуры мышечных волокон и некротические изменения в сердце при стрессе объясняют нарушением мембранного транспорта кальция, ибо удаление кальция из миофибрилл - необходимый процесс нормального расслабления. Основу указанного нарушения составляет повышение проницаемости мембран саркоплазматического ретикулума для кальция и снижение активности фермента Са-АТФ-азы. После перенесенного стресса выявлено снижение адренореактивности сердечной мышцы. По Ф.З. Меерсону, патогенез повреждений сердечной мышцы при стрессе можно представить следующим образом: высокие концентрации катехоламинов ^ активация перекисного окисления липидов и накопление перекисных соединений ^ лабилизация лизосом ^ повреждение мембран перекиси липидов и протеолитическими ферментами, мембран сарколеммы и саркоплазматического ретикулума ^ нарушение транспорта кальция в миокардиальных клетках ^ кальциевая контрактура и гибель клеток.
Стресс является также важным инициальным моментом формирования гипертонической болезни вследствие активации симпато- адреналовой и гипоталамо-гипофизарно-надпочечниковой систем и последующего расстройства водно-солевого обмена и сосудистого тонуса.
Таким образом, уже на примере расстройств сердечно-сосудистой системы мы видим, как стресс-синдром превращается из звена адаптации в звено патогенеза неинфекционных заболеваний.
Изменения крови при стрессе. Изменения крови и их механизмы при однократном и повторном стрессе (иммобилизация, раздражение электрическим током, мышечная нагрузка, гипоксия, кровопотеря, введение эритропоэтинов и др.) подробно изучены П.Д. Го- ризонтовым с соавт. Продолжительность, интенсивность изменений крови и развитие всех стадий стресса определяются длительностью и специфичностью действующего на организм стрессора. Важные с точки зрения теории и практики медицины факты1 были получены исследователями благодаря комплексному изучению различных отделов системы крови (лимфоидных органов, периферической крови, костного мозга), что позволило судить о реакциях системы крови как единого органа. Ими установлены два периода изменений в течение 48-72 часов от начала воздействий.
В первом периоде, продолжительностью 12 часов, в крови обнаруживается нейтрофилез, лимфо- и эозинопения, уменьшение числа клеток в лимфоидных органах. В костном мозге отмечено уменьшение количества зрелых нейтрофильных гранулоцитов, преходящее увеличение содержания лимфоцитов.
К концу первых суток изменения в крови нивелировались и начинался второй период, формирование которого определяется спецификой примененного стрессора. Изменения происходят в основном в костном мозге в виде активации эритро- и лейкопоэза, явлений гиперплазии, снижения количества лимфоцитов (как Т-, так и В- лимфоцитов). В селезенке количество лимфоцитов нормализуется, а в тимусе продолжается снижение числа клеток.
Такие закономерности имеют место у разных видов животных (мыши, крысы, морские свинки).
Анализ подобных изменений в зависимости от возраста показал, что только через 1 месяц после рождения изменения крови соответствуют сдвигам, наблюдаемым у взрослых животных. Особенно это касается лимфопении, уменьшения клеток в тимусе и лимфоидного пика костного мозга. Эти изменения крови характеризуют первую стадию стресса - реакцию тревоги.
По мнению П.Д. Горизонтова и соавт. с избыточной продукцией и секрецией гормонов глюкокортикоидов связаны эозино- и лимфопения, снижение клеток в тимусе, накопление гемопоэтических клеток в первом периоде стресса и гранулоцитопоэз - во втором периоде. Такие же изменения, как нейтрофильный лейкоцитоз, лимфоидный пик в костном мозге, а также уменьшение лимфоидных клеток в селезенке не зависят от гормональных влияний.
Основное значение в опустошении лимфоидных органов принадлежит миграции клеток из этих структур; снижению пролиферативной активности и распаду лимфоцитов в этих органах принадлежит меньшая роль, хотя при некоторых стрессорных воздействиях (например, гипоксии) распад клеток — основная причина лимфопении.
Показано также различие механизмов миграции лимфоцитов при стрессе из тимуса и селезенки. Мобилизация клеток из тимуса обусловлена действием избытка гормонов гипофизарно- адренокортикальной системы, а в селезенке - повышением тонуса гладкой мускулатуры в результате возбуждения «-адренорецепторов. Сокращение гладкой мускулатуры способствует выбросу в кровь большого числа лимфоцитов.
Причиной лимфопении является увеличение выхода их из крови и поступление в ткани, особенно в костный мозг. Накопление лимфоцитов в костном мозге в стадии тревоги, по мнению П.Д. Горизонтова с соавт. имеет большое биологическое значение, так как увеличивает его иммунокомпетентность.
Через 1-3 суток после однократного стрессорного воздействия регистрируется период повышенной резистентности, и повторное воздействие приводило в течение первых шести дней только к изменениям со стороны периферической крови.
Таким образом, при повторном однократном действии стрессорного фактора в организме возникает ответ меньшей степени выраженности в виде изменений крови, но без реакции со стороны кроветворных органов, что необходимо рассматривать как вторую стадию стресса - стадию резистентности.
В третьей стадии стресса, возникающей в результате сильного и продолжительного действия стрессоров, формируется стадия истощения, для которой характерно снижение числа клеток в различных отделах системы крови до величин, несовместимых с жизнью.
Влияние стресса на иммунитет. В стадии тревоги в зависимости от силы и длительности действия стрессора и, особенно в условиях действия экстремальных факторов отмечается торможение иммунобиологических механизмов, следствием чего обычно является уменьшение интенсивности аллергических реакций, снижение резистентности к опухолевому росту, повышение чувствительности к вирусным и бактериальным инфекциям.
В основе иммунодепрессии лежат увеличение концентрации глюкокортикоидных гормонов и возникающее вследствие этого перераспределение клеток, торможение митоза лимфоцитов, активация Т- супрессоров, цитолический эффект в тимусе и лимфоидных узлах. Иммунодепрессия характерна как для гуморальной, так и клеточной форм иммунитета.
В стадии резистентности регистрируется не только восстановление, но и увеличение иммунитета.
Если интенсивность и продолжительность стрессора очень велики, восстановления, а тем более стимуляции иммунитета не происходит и, по мнению П.Д. Горизонтова с соавт. формируется третья фаза стресса, проявляющаяся формированием вторичной иммунологической недостаточности.
Нарушения обмена веществ при стрессе. Усиленная продукция катехоламинов при стрессе активирует фосфорилазу печени и распад гликогена в этом органе. Кроме того, избыток глюкокортикоидов стимулирует в печени и почках глюконеогенез. Эти два механизма объясняют важное проявление стресса - гипергликемию, что увеличивает образование и инкрецию инсулина. Поэтому в условиях длительного стресса вследствие постоянной и продолжительной гипергликемии, и стимуляции β-клеток островкового аппарата поджелудочной железы может наступать напряжение, перенапряжение и истощение инсулярного аппарата, что и составляет основу механизма сахарного диабета при стрессе.
Иногда он называется диабетом напряжения.В стадии истощения имеет место снижение содержания глюкозы в крови вследствие отсутствия запасов гликогена в печени. Так, в экспериментах на крысах показано, что в условиях 24-часового голодания в печени крыс обнаруживаются следы гликогена.
В условиях стресса ингибируется гликолиз в печени, мышцах, сердце, не изменяется в мозге и активируется в надпочечниках (Л.Е. Панин). Это связано с изменением активности основных ферментов гликолиза - гексокиназы и фосфорилазы печени.
Глюконеогенез в печени и почках (т.е. синтез глюкозы из неуглеводистых продуктов - пирувата, лактата, глюкогенных аминокислот) осуществляется с участием ключевого фермента фосфоэнолпируват- карбоксиназы и резко возрастает при стрессе.
Активации глюконеогенеза способствует снижении инсулина в крови, особенно в стадии резистентности, что за счет активаций контринсулярных гормонов обеспечивает мобилизацию жира, ингабиро- вание гликолиза и усиление глюконеогенеза. Кроме того, это обеспечивает переключение энергетического обмена на липидный. Именно в этот период, по мнению Л.Е. Панина, источником углеводов становится глюконеогенез, основу которого составляют глюкогенные аминокислоты: частично гликоген в печени образуется из лактата через цикл Кори. Именно в этот период основным энергетическим материалом становятся жирные кислоты, а их продукты - кетоновые тела - как энергетический материал окисляются в мозге, почках, сердце, мышцах. Интенсивно используются жирные кислоты, особенно в мышцах.
Как показывают клинические наблюдения, при стрессе снижается чувствительность нервной ткани к дефициту углеводов, так как в биоэнергетике возрастает роль кетоновых тел, образующихся за счет интенсивного использования жирных кислот в качестве энергетического материала.
По данным Л.Е. Панина, дефицит углеводов при стрессе начинает сказываться в стадии истощения, что проявляется в дальнейшей активации симпато-адреналовой системы и выбросе инсулина, но к этому времени углеводные резервы исчерпываются полностью. Поэтому в стадии истощения развивается гипогликемия, которая ведет к гибели организма вследствие невозможности энергообеспечения.
В результате избыточной продукции катехоламинов и глюкокортикоидов имеет место усиленная мобилизация жиров из жировых депо с формированием гиперлипидемии и особенно гиперхолестеринемии, что способствует отложению холестерина в сосудах и развитию атеросклероза. Клинические наблюдения показывают увеличение в крови при стрессе общих липидов, общего холестерина, свободных жирных кислот, суммарной фракции липопротеинов низкой плотности. При стрессе усиливается перекисное окисление липидов, и образующиеся перекиси вызывают прямое повреждение сосудистой стенки. Доказательством того, что происходит повреждение клеточных мембран, является выраженное увеличение количества ферментов в крови.
В эксперименте получен атеросклероз путем назначения животным безантиоксидантной диеты, содержащей избыток перекисей липидов. В этом случае, по данным Ф.З. Меерсона, перекисями повреждаются сосуды с отложением в них кальция и липидов. Этот процесс ускоряется в условиях иммобилизационного стресса и тормозится ингибитором окислительных процессов — ионолом.
Таким образом, стресс может усиливать и способствовать формированию атеросклероза за счет формирования стрессорной гиперлипидемии и особенно гиперхолестеринемии, а также повреждения мембран клеток перекисями липидов.
Как уже говорилось, в условиях стресса возрастает роль липидов в биоэнергетике организма, и энергетический обмен переключается с углеводов на липиды, что находит отражение и в перестройке дыхательной цепи в митохондриях клеток. Это проявляется в уменьшении образования ацетил-КоА из углеводов и увеличением образования его из жирных кислот.
Первый путь окисления углеводов и липидов через цикл Кребса был назван Л.Е. Паниным «углеводным», второй — в виде фосфори- лирующего окисления липидов по перекисному механизму, назван «липидным».
Полагают, что в стадии резистентности энергетический обмен с углеводного типа переключается на липидный, а цАМФ является тем медиатором, с помощью которого происходит переключение энергетического обмена. Увеличение цАМФ в тканях (печень, мышцы) тормозит гликолиз за счет ингибирования гексокиназы. Подавляется липогенез и активируется липолиз. В митохондриях, особенно печени, возрастает скорость фосфорилирующего окисления как углеводных (пируват), так и, особенно, липидных субстратов (Л.Е. Панин).